Tell me the whole story: the role of product labelling in building user confidence in biosimilars in Europe
Published on 2014/09/02
Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(4):188-92.
|
Abstract: |
Submitted: 1 September 2014; Revised: 23 September 2014; Accepted: 9 October 2014; Published online first: 22 October 2014
Introduction
Europe has been instrumental in the global development of biosimilars, with 12 biosimilar molecules approved, marketed as 18 brands in six classes: somatropin, epoetins, filgrastim, follitropin alfa, insulin glargine, as well as the first biosimilar monoclonal antibody (mAb), infliximab (marketed as Inflectra, Remsima), approved in September 2013 [1, 2].
The requirements and procedures for the marketing authorization for medicinal products for human use in Europe Union (EU) are primarily laid down in Directive 2001/83/EC, as amended. This Directive provides distinct legal frameworks for innovator products, small molecule generic products and biosimilar products, dictating the amount of quality, preclinical and clinical data required to support approval and marketing. To the latter point, small molecule generic products whose formulation or manufacturing method have not been modified in any way that may impact the bioavailability [3] do not require any preclinical or clinical trial data of their own as bioequivalence with a reference product (innovator) can be established through in vitro testing alone. For biosimilar products, in contrast, such a formulaic approach cannot be used. The amount of data to be generated is determined in a stepwise approach, directly comparing the biosimilar with the reference product and the outcome informing the next step. This may result in the need for more or less extensive comparative clinical data. Indeed, the European Medicines Agency (EMA) draft revised guideline, ‘Guideline on Similar Biological Medicinal Products’ states that ‘standard generic approach (demonstration of bioequivalence with a reference medicinal product by appropriate bioavailability studies) which is applicable to most chemically-derived medicinal products is in principle not appropriate to biological/biotechnology-derived products due to their complexity’ [4].
The legal framework for biosimilars enabled EMA to pioneer the regulatory review of biosimilars according to a new scientific approach [5]. However, legal and scientific distinction has not been consistently reflected in the product labelling, with there being differences in the level of detail provided.
The Committee for Medicinal Products for Human Use (CHMP) appears to have adopted a labelling approach used for small molecule generic products, with only a small distinction made between the product labelling of the reference and generic products for biosimilar products. This is also illustrated with the recent EU approval(s) of Remsima and Inflectra [1, 2], where such an approach was applied.
Product labelling: what every physician needs to know
As defined by EMA [6] the product labelling, in particular the Summary of Product Characteristics (SmPC) is a key component of the marketing authorization of all medicines in the EU and the basis of information for healthcare professionals on how to use a medicine safely and effectively. Updates to the SmPC are made throughout the lifecycle of a medicine as new efficacy and/or safety data emerge or change in the safety profile become apparent. The SmPC also forms the basis for the preparation of the Patient Information Leaflet (PIL), which is an important document for relaying information on medicines to patients.
Instructions on what needs to be detailed in the SmPC are provided in the ‘Guideline on Summary of Product Characteristics’ and when read in conjunction with other guidelines clearly defines the level and type of information that should be reflected in SmPC for all products, including biosimilar medicines. Despite there being general guidance pertaining to the content required for the SmPC as discussed, it is acknowledged there is a requirement for further dialogue regarding detailed specific guidance for biosimilars to be developed.
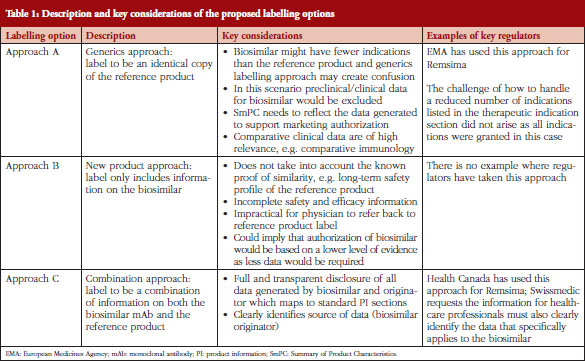
The structure of the SmPC in relation to biosimilars was addressed by the chair and members of the Biosimilar Medicinal Products Working Party (BMWP), Schneider et al. [7] using mAb as the example. The BMWP authors outlined three potential labelling scenarios, see Table 1, to be considered for labelling of biosimilars, ranging from an identical product label to the reference mAb through to a completely distinct label only reflecting data generated for the biosimilar [7]. As each of the labelling scenarios presented had their pros and cons, the BMWP authors were of the opinion that it was not yet clear how best to conclude the matter.
Other groups have also sought to address the issue, including the European Biopharmaceutical Enterprises (EBE) [8], EuropaBio [9] and the Association of the British Pharmaceutical Industry (ABPI) [10].
The EBE welcomes the BMWP’s acknowledgement that adequate scientific information for biosimilars should be provided and in this regard the product label is the essential component for prescribers [7]. This also corresponds with the key learnings from the Process on Corporate Responsibility in the field of Pharmaceuticals, initiated by DG Enterprise that physician perception and patient acceptance impacts biosimilar uptake [11]. It was concluded that a robust regulatory framework, effective risk management, transparency, and continued education would help engender confidence in the appropriate use of innovator biological medicines and biosimilars. To explain their rationale for not distinguishing the reference from the biosimilar label, the BMWP of EMA has emphasized concern that differential product labelling could implicitly suggest a difference between the biosimilar and the reference product. The latter could be prone to misperception by prescribers who could falsely conclude that the level of evidence created to lead to approval of a biosimilar is less than the usually expected standards for a novel product, if the concept of biosimilars is not understood [1].
While the EBE [12] acknowledges the reasons and concerns highlighted by BMWP, the EBE position is that the SmPC would be better served if it were represented by a combination of information from both the biosimilar and the reference product, see Table 1, if the label is different. Such an approach is of crucial importance in developing an understanding and acceptance of biosimilars.
Labelling standard of biosimilars: why a combined approach fits the purpose
Three possible scenarios for the labelling (including SmPC and PIL) of biosimilars have been described by Schneider et al. [7]. Adaptations of the proposed labelling options of biosimilars are briefly described in Table 1, which gives an overview of the approaches and relevant key considerations:
EBE believes that Approach C is best suited. First of all, the unique considerations that apply to biosimilars (as compared to generics) exclude Approach A as a suitable option because it does not include relevant data the biosimilar manufacturer has compiled for its clinical comparison. Yet prescribers may want to see such biosimilar data alongside that for the originator, as this will explain the basis for which indications have been approved. Furthermore, by using biosimilar data only (Approach B) characterization data is not included, thus neglecting the proof of biosimilarity. This characterization assumes that the long-term safety profile for the reference product should be applied to the biosimilar and that therefore class warnings, etc.; are appropriate for both products. In addition, because Approach B only provides biosimilar data, this also means that the prescriber needs to refer back to the reference product’s label to complete their understanding of the product, and this is not practical. Approach C is the more balanced approach that can enable transparent disclosure of all relevant information related to the biosimilar and the reference product. Furthermore, Approach C is an approach that also allows transparency on where the data generated comes from, either from the originator or from the biosimilar developer and, additionally, which indications are granted by extrapolation.
Five points that show the need for detailed specific guidance on what a transparent label for biosimilars would look like
EBE identifies that there are five important points for consideration, which arise when prescribers refer to the SmPC of a biosimilar, and these should be considered in any policy guidance related to the matter. They are:
- Quantity of data. There is the concern that physicians would directly relate similarity to the amount of clinical data provided in the SmPC. However, in accordance with the biosimilar concept the more similar the product is to its reference product, the less clinical data will need to be generated during its own development. Therefore, only by the SmPC containing a combination of information/data relevant to the originator and biosimilar could the CHMP conclusions be provided to the prescriber. Some stakeholders also argue that the inclusion of preclinical and clinical data on the biosimilar is misleading because the assessment of biosimilarity is primarily based on extensive analytical comparisons which may not be included in the SmPC. EBE is of the opinion that biosimilars must be labelled according to agreed guidelines that apply to all medicinal products. However, since the SmPC does not usually contain analytical data, it would also be important to explain the inclusion of relevant analytical data to prescribers.
- Extrapolation of indication. In terms of labelling, taking a generics approach (the identical product label) could lead to misperceptions by physicians that these are identical products, which biosimilars cannot be. As Weise et al. [13] state in their article titled Biosimilars: what clinicians should know: ‘It must be clearly understood that a biosimilar, as opposed to a small chemical generic, cannot automatically claim all indications of the reference product and that any extrapolation of data requires sound scientific justification’. Applying a generics style of labelling to biosimilars may lead physicians to the (wrong) assumption that the generics approach would also apply to extrapolation of indications. In contrast, transparent information about the decision-making would not only facilitate the understanding of the nature of biosimilars (and their assessment by CHMP) but also increase confidence in their use.
- Switching. Decisions around switching require transparent product information so that prescribers can make their choices confidently. As the EMA Q&A document on biosimilar medicines [14] states the following about switching: ‘For questions related to switching from one biological medicine to another, patients should speak to their doctor and pharmacist’, it is therefore of upmost importance that there is full transparency that the product in question has been licensed as a biosimilar and its terms of approval are explained in the SmPC and PIL to enable an informed decision by all end-users. There should be a debate about to what extent switching data, where available, should be included on the product label.
- Pharmacovigilance. In supporting the aims of the Pharmacovigilance Directive reporting requirements for biologicals (including brand name and batch number) should be included on the prescription in order to improve traceability and enhance pharmacovigilance. Some biological products already include guidance of this sort in Section 4.4 (Special warnings and precaution for use) of their SmPC, see for example the SmPC for adalimumab – in order to improve traceability of biological medicinal products, the trade name and the batch number of the administered product should be clearly recorded [15].
- Drift. Once a biosimilar has been approved, there is currently no legal or regulatory requirement for biosimilarilarity to be re-established at any time. It is well acknowledged that the reference product and its biosimilar will have separate life-cycles which could affect the safety and efficacy profile, while having no change to the other, and which could undergo patterns of drift and evolution that ultimately result in two products that are no longer biosimilar (divergence) [16].
In light of the above considerations, EBE maintains that a single sentence added in section 5.1: Pharmacodynamic properties of the SmPC and a reference to the EMA homepage informing the prescriber that the product is a biosimilar and providing reference for further information does not seem to be sufficient. The information refers the reader to the EMA homepage [17], offering no guidance to physicians and patients to help them navigate to the relevant information and documentation.
Besides this, EBE considers that section 5.1 is not the most relevant section for such a statement, as the information applies to the whole product label and not only to pharmacodynamic properties. More importantly, details of pharmacodynamic properties are not reflected in the PIL and there is a lack of transparency informing patients to the fact a product is a biosimilar medicine.
The SmPC and other supporting documents
It is acknowledged that the SmPC is the most widely used reference document for physicians, however, there are other important documents published in the EU which provide information about the product and its basis for approval: the European Public Assessment Report (EPAR) which is a summary of the review and conclusions of the scientific assessment by CHMP; and in a patient friendly format: the Patient Information Leaflet. Table 2 below provides a short description of each of these documents.

For recent biosimilar approvals the ‘generic’ approach to labelling has been agreed by EMA and therefore alludes that it has been considered as representing the most appropriate method for communicating information to physicians. While it is acknowledged that the ‘generic’ labelling approach, taken for the recently approved biosimilars, infers that physicians who would like to have an in-depth understanding of the scientific discussion can refer to the published EPAR on their website, it should be noted that these documents have important limitations in their use as vehicles for educating physicians and patients across the EU, see Table 2.
As the full EPAR information is only available in English, this effectively restricts the number of physicians across the EU who can rely on it as a source of information. In practice, the prescriber would need to refer to separate EPARs, the biosimilar(s) and reference product, in order to scrutinise the data generated when deciding whether to switch their existing medication to the biosimilar. If more than one biosimilar product were available, this would further increase the time needed by a physician to evaluate the therapeutic information. Physicians are already dissatisfied with the increasing time they must spend on administrative tasks and paperwork, as this limits their time for face-to-face patient care [18].
EPARs are primarily designed to provide information on how a medicine was assessed by CHMP and to describe scientific conclusions of the relevant Agency committee. Physicians are likely to be unaware that the EPAR document itself is not updated, but rather is complemented by additional documents such as summaries called ‘Procedural steps taken and scientific information after authorisation’ [19].
Recently, the Alliance for Safe Biologic Medicines (ASBM) [20] surveyed 470 prescribers located in France, Germany, Italy, Spain and the UK regarding information sources used for learning about medicines. The respondents were specialists who prescribed biologicals, including nephrologists, rheumatologists, dermatologists, neurologists, endocrinologists and oncologists and so their perspectives reflected hands-on clinical experience with biologicals in a therapeutic setting. The results indicate that among available information sources, EPAR was the least preferred method (19%), while the SmPC was the next most important source document physicians refer to when they need specific details about a product (43%) after published literature.
Detailed biosimilar labelling guidance in other jurisdictions
Whilst EMA, pioneered the biosimilar approval pathway, specific biosimilar product labelling guidelines have as yet to be developed, whereas other countries have already made provisions on how biosimilar product labels should be presented.
When reviewing the situation in the US, Switzerland and Canada, not unexpectedly, it becomes evident that guidance on the content of biosimilar product labels varies from country to country.
United States
In the US, although biosimilars are still to be registered, FDA has recognized the importance of labelling for biosimilars and is working to reduce potential ambiguities by ensuring clear statements within appropriate sections of the Prescribing Information [21]. Section VIII of the FDA draft guidance on scientific consideration in demonstrating biosimilarity states that labelling of a proposed product should include all the information necessary for a healthcare professional to make prescribing decisions, including a clear statement advising that the product approved is a biosimilar and that it has or has not been determined interchangeable with the reference products [22].
Switzerland
In Switzerland, Swissmedic has also been clear in differentiating between the terms ‘reference product’ and ‘comparator product’ in order to emphasise the fact that candidate biosimilars are characterized by documentary reference to the Swiss reference product [23]. The guideline also lays out the responsibilities to keep the PI of the biosimilar up to date is that of the authorization holder. In particular, the authorization holder must actively monitor changes to the safety text of the reference product and must submit either an appropriate application for a variation or provide clear scientific justification if the texts are not to be adapted.
Canada
Health Canada’s approach for their first approved biosimilar mAb (Remsima/Inflectra) approved in January 2014 shows the clearest distinction to the approach that EMA followed, see Table 1. As the guideline states, the sponsor of a biosimilar is not able to utilize the product monograph of the reference biological drug in its entirety as that of its own product [24]. Furthermore, the monograph details additional requirements clarifying that the product is a biosimilar. These include key data on which the decision for market authorization was made, tables showing the results of the comparisons between the biosimilar and reference biological drug, and information on the indications approved for use.
The way forward
On the basis of the arguments provided, EBE recommends that an approach of greater transparency within the product labelling of the biosimilar be provided. A combination of information on both the biosimilar and the reference product (Approach C) is recommended and to define what this entails, greater dialogue is required between EMA, BMWP, industry, opinion leaders and patients. As EPAR is not a preferred information source, in its current setting, it should not replace the important role of SmPC as the primary point of reference.
EBE is of the position that the generation of detailed specific guidance on the product labelling of biosimilars is of crucial importance to realize consistency and transparency of biosimilar labels, which will lead to a better understanding and acceptance of these products with all stakeholders. Most importantly, consideration should be given to ensuring the SmPC for biosimilars details information most relevant to the prescriber.
Although it will be challenging to achieve an ideal solution which encompasses the needs of all stakeholders, it is preferable that clinicians have ready access to the relevant information regarding biosimilars to enable informed decision-making by physicians and patients and therefore ensures safe and effective use of the medicine. Clearly, further education and dialogue on biosimilar concepts is needed and this is a widely shared agenda. Equally important is the need to build trust together with understanding, and the foundation for this is transparency and open dialogue. To meet these aims, EBE recommends a thorough consultation with all stakeholders to explore the needs and the best approaches for providing appropriate product labelling guidance for biosimilars in the EU.
Competing interests: This paper is authored and funded by the European Biopharmaceutical Enterprises (EBE) and represents the policies of the organization.
EBE is the European trade association that represents biopharmaceutical companies of all sizes operating in Europe. Membership is open to all companies using biotechnology to discover, develop and bring new medicinal products to market.
Provenance and peer review: Not commissioned; externally peer reviewed.
Contributing authors from EBE
Keith Watson, AbbVie
Virginia Acha, Catherine Akers, Amgen
Alexander Roediger, MSD
Åsa Rembratt, Novo Nordisk
Edward Hume, Pfizer
Fabio Bisordi, Marloes van Bruggen, Roche
References
1. European Medicines Agency. European public assessment reports: Inflectra. 2013 [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002778/human_med_001677.jsp&mid=WC0b01ac058001d124
2. European Medicines Agency. European public assessment reports: Remsima. 2013 [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002576/human_med_001682.jsp
3. European Medicines Agency. Committee for Medicinal Products for Human Use. Guideline on the investigation of bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1/Corr++. 20 January 2010 [homepage on the Internet]. 2010 Mar 10 [cited 2014 Sep 23]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
4. European Medicines Agency. Guideline on similar biological medicinal products. CHMP/437/04 Rev 1. 22 May 2013 [homepage on the Internet]. 2013 Jun 19 [cited 2014 Sep 23]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/05/WC500142978.pdf
5. European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. EMA/CHMP/BMWP/403543/2010. 30 May 2012 [homepage on the Internet]. 2012 Jun 11 [cited 2014 Sep 23]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf
6. European Medicines Agency. How to prepare and review a summary of product characteristics [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000357.jsp&mid=WC0b01ac05806361e1
7. Schneider CK, Vleminckx C, Gravanis I, Ehmann F, Trouvin JH, Weise M, et al. Setting the stage for biosimilar monoclonal antibodies. Nat Biotechnol. 2012;30(12):1179-85.
8. European Biopharmaceutical Enterprises. EBE position paper biosimilars labeling. Summary of Product Information Characteristics and Patient Information Leaflet. 21 April 2013 [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://www.ebe-biopharma.eu/documents/59/66/EBE-position-paper-on-Biosimilars-Labelling
9. EuropaBio. Statement on labelling of biosimilars. 15 September 2014 [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://www.europabio.org/positions/europabio-statement-labelling-biosimilars
10. ABPI. ABPI position on biosimilar medicines. February 2013. Available from: http://www.abpi.org.uk/our-work/library/industry/Documents/ABPI%20biosimilars%20position%20paper.pdf
11. European Commission. A consensus information document. What you need to know about biosimilars. April 2013 [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://ec.europa.eu/enterprise/sectors/healthcare/files/docs/biosimilars_report_en.pdf
12. European Biopharmaceutical Enterprises. Biosimilars [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://www.ebe-biopharma.eu/priorities/biosimilars
13. Weise M, Bielsky MC, De Smet C, Ehmann F, Ekman N, Giezen J, et al. Biosimilars: what clinicians should know. Blood. 2012;120(26):5111-7.
14. European Medicines Agency. Questions and answers on biosimilar medicines (similar biological medicinal products) 2012 [homepage on the Internet]. 2012 Sep 27 [cited 2014 Sep 23]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/12/WC500020062.pdf
15. European Commission. Directive 2010/84/EU of the European Parliament and of the Council of 15 December 2010 amending, as regards of pharmacovigilance, Directive 2001/83/EC on the Community code relating to medicinal products for human use [homepage on the Internet]. 2010 Dec 31 [cited 2014 Sep 23]. Available from: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:348:0074:0099:EN:PDF
16. Ramanan S, Grampp G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs. 2014;28(4):363-72.
17. European Medicines Agency [homepage on the Internet]. [cited 2014 Oct 9]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/home/Home_Page.jsp&mid=
18. NHS Confederation. Challenging bureaucracy. 2013 [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://www.nhsconfed.org/Publications/reports/Pages/challenging-bureaucracy.aspx
19. European Medicines Agency. European public assessment reports [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages%2Fmedicines%2Flanding%2Fepar_search.jsp&murl=menus%2Fmedicines%2Fmedicines.jsp&mid=WC0b01ac058001d124&searchTab=searchByAuthType&alreadyLoaded=true&isNewQuery=true&status=Authorised&status=Withdrawn&status=Suspended&status=Refused&keyword=Enter+keywords&searchType=name&taxonomyPath=&treeNumber=&searchGenericType=biosimilars&genericsKeywordSearch=Submit
20. Safe Biologics. Alliance for Safe Biologic Medicines (ASBM). ASBM shares EU survey results at Paris media briefing. 10 June 2014 [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://safebiologics.org/recent-events.php
21. Davis C, Bowker GM. Labeling standards for biosimilar products. Therapeutic Innovation & Regulatory Science. 2014;48(3):367-70.
22. U.S. Food and Drug Administration. Draft guidance for industry on scientific considerations in demonstrating biosimilarity to a reference product. Rockville, MD. February 2012 [homepage on the Internet]. 2012 Feb 8 [cited 2014 Sep 23]. Available from http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
23. Swissmedic. Swiss Agency for Therapeutic Products. Questions and answers concerning the authorisation of similar biological medicinal products (biosimilars). 17 February 2014 [homepage on the Internet]. [cited 2014 Sep 23]. Available from: https://www.swissmedic.ch/ueber/00134/00519/01933/index.html?lang=en
24. Health Canada. Guidance for sponsors: information and submission requirements for Subsequent Entry Biologics (SEBS). 2010 [homepage on the Internet]. [cited 2014 Sep 23]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/applic-demande/guides/seb-pbu/seb-pbu_2010-eng.php
|
Author for correspondence: Edward Hume, Pfi zer Ltd, Walton Oaks, Dorking Road, Tadworth, Surrey, KT20 7NS, UK |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



One Reply to “Tell me the whole story: the role of product labelling in building user confidence in biosimilars in Europe”