The EU is ready for non-biological complex medicinal products
Published on 2016/04/07
Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(1):30-5.
|
Abstract: |
Submitted: 15 March 2016; Revised: 1 April 2016; Accepted: 4 April 2016; Published online first: 15 April 2016
Non-biological complex drugs (NBCDs) are defined scientifically as not being a biological medicinal product where the active substance is not a homo-molecular structure, but consists of different closely related and often nanoparticulate structures that cannot be isolated and fully quantitated, characterized and/or described by physicochemical analytical means, where the structural elements that might impact the therapeutical performance are unknown [1]. Nanomedicines, such as liposomes, polymeric micelles, glatiramoids, iron-carbohydrate complexes and nanocrystals, as well as dry powder inhalers are examples of NBCDs. Although the definition of NBCD is not officially recognized and there is no corresponding term in European Union (EU) pharmaceutical legislation or scientific guidance, it will be argued in this paper that the existing EU legislation and guidance is equipped to adequately assess the quality, safety and efficacy, as well as the lifecycle management, of such a group of medicinal products. We will describe the different mechanisms through which the European Medicines Agency (EMA) promotes the development and entry into the market of innovative medicinal products. This paper aims at addressing important issues, such as the need to follow the biosimilar approach for comparability, harmonization of technical requirements across regions or preparedness of EU legislation for NBCDs, raised in GaBI on the regulation of NBCDs, particularly those mentioned in the paper entitled ‘Non-Biological Complex Drugs and their follow-on versions: time for an editorial section’ published online first on 2 November 2015 [2]. We will start by detailing the role of EMA, the scope of the centralized procedure and the legal basis of submission of marketing authorization procedures. We will continue by providing some examples of products that would fall under the author’s definition of NBCD and describing how these can be submitted for a marketing authorization application in the EU under the existing legislation and scientifically assessed under the framework provided by the existing scientific guidance. We will conclude by substantiating how the EU is adequately prepared for NBCD.
Marketing authorization procedures and legal basis of submission
EMA is an EU body established in 1995 in accordance with Council Regulation (EEC) No 2309/93, now superseded by Council Regulation (EEC) No 726/20014. EMA is responsible for the coordination of the existing scientific resources from a network of national experts for the evaluation, supervision and pharmacovigilance of medicinal products. As part of the mission of EMA, the agency also supports research and innovation to stimulate the development of new medicines to address public health needs. One of the main responsibilities of the agency is the scientific evaluation of applications for EU marketing authorizations for human and veterinary medicines in the centralized procedure. The centralized procedure is compulsory for a discrete class of products:
- Medicinal products developed by recombinant DNA, controlled expression of genes coding for biotechnologically active proteins in prokaryotes and eukaryotes including transformed mammalian cells, hybridoma and monoclonal antibody methods
- Medicinal products for human use containing a new active substance for the treatment of acquired immune deficiency syndrome, cancer, neurodegenerative disorder, diabetes, autoimmune diseases and other immune dysfunctions, viral diseases
- Designated orphan medicinal products
Applicants still have the opportunity to apply for a marketing authorization through the centralized procedure if the medicinal product contains a new active substance not authorized in the EU; or if it constitutes a significant therapeutic, scientific or technical innovation or that the granting of the authorization is in the interest of patient health at Union level. A medicinal product that would not fall under the mandatory scope may be authorized through the EU via the centralized procedure or the national procedures managed by the National Competent Authorities of each Member State of the EU. The Directive 2001/83/EC [3] sets the requirements to support a marketing authorization application for medicinal products relating to medicinal products for human use and is equally applicable regardless of the procedure through which the application is made. Similarly the scientific guidance is drafted and adhered to by the same group of national experts responsible for assessing the quality, safety and efficacy of medicines submitted to the centralized, decentralized, mutual recognition or purely national procedures. The Co-ordination Group for Mutual Recognition and Decentralised Procedures – Human (CMDh) for human medicinal products [4] also consults the Committee for Medicinal Products for Human Use (CHMP) and its working parties for scientific opinions when necessary. As a result, all medicines approved in the EU comply with the same legal requirements and are reviewed considering equal scientific standards.
The extent (e.g. full versus bridging studies) and nature (e.g. new active substance versus comparative versus well-established use) will determine the options under which a marketing authorization application may be made.
The legal basis for submission, as detailed in Directive 2001/83/EC, are:
- Stand-alone application comprised of a full set of pharmaceutical, preclinical and clinical data, or mixed applications comprised of a full set of pharmaceutical data with the option of a combination of reports of limited non-clinical and/or clinical data – Article 8(3)
- Generic medicinal product – Article 10(1)
- Hybrid medicinal product – Article 10(3)
- Similar biological medicinal product or biosimilar – Article 10(4)
- Well-established use application, where preclinical and clinical data is replaced by detailed references to published scientific literature if it can be demonstrated that the active substance(s) has been in well-established medical use within the EU for at least 10 years, with recognized efficacy and acceptable safety – Article 10(a)
- Fixed-combination application, where more than one active substance is used in combination in the same medicinal product for therapeutical purposes – Article 10(b)
- Informed consent applications, where the holder of an authorized medicinal product allows the quality, non-clinical and clinical data submitted for its authorization to be used for future applications – Article 10(c)
NBCDs can potentially be submitted under the seven legal basis above, even as a biosimilar since the determining factor is the biological* nature of the reference medicinal product, regardless of the non-biological nature of the manufacturing process of the NBCD being submitted for a marketing authorization.
It has been debated in this journal [2] that the current EU pharmaceutical legislation does not have a dedicated regulatory scheme that takes into account the special nature of NBCD follow-on products. There exist two options in legislation for a follow-on medicinal product of an NBCD. Depending on the extent of data needed to support bioequivalence, a follow-on of an NBCD will either fall under one of the following legal basis:
- Generic, if the follow-on medicinal product has the same qualitative and quantitative composition in active substance(s) and the same pharmaceutical form as the reference medicinal product, and whose bioequivalence with the reference medicinal product has been demonstrated by appropriate bioavailability studies.
- Hybrid, where the bioequivalence cannot be demonstrated through bioavailability studies or in case of changes in the active substance(s), therapeutic indications, strength, pharmaceutical form or route of administration, vis-à-vis the reference medicinal product, the results of the appropriate preclinical tests or clinical trials shall be provided.
In the scientific reflection papers and guidance published, EU regulators are open to the possibility of quality data being sufficient to demonstrate the bioequivalence of two NBCDs [5], it is however recognized that differences observed when performing pharmaceutical comparability studies between reference and test product, or during the different stages of the development of a medicinal product, may require in vivo studies to demonstrate bioequivalence. The extent of in vivo studies that may be necessary to support bioequivalence are considered on a case-by-case basis according to the complexity of the formulation and the data gathered cumulatively by the different quality, non-clinical and clinical comparative studies. Furthermore, it is also recognized that only certain aspects of the conventional bioequivalence approach [6] are applicable and in some cases the need to provide clinical studies to demonstrate the safety and efficacy of follow-on products may be justified. The body of evidence obtained in the quality, non-clinical and clinical purposely-designed studies for NBCDs will be considered as a whole when assessing the benefit risk of the medicinal product undergoing an application for a marketing authorization.
This approach allows for data requirements to be adapted according to the nature of the product, differences observed during the comparability studies and claimed therapeutic indications, hence, guaranteeing the demands from regulators are tailored to a specific product or class of products.
As defended by Schellekens and Borchard [7], NBCDs are made of structures that cannot be isolated and fully quantitated, characterized, and/or described by analytical means, to which it is unknown which therapeutic elements might impact therapeutic performance and with clinical performance highly dependent on the production process. Schellekens and Borchard have argued that an approach common to that applied for biosimilars should also be considered for follow-on NBCDs, it is therefore relevant to identify what are the differences between the two approaches. Although by definition Article 10(4) – biosimilar applications – is not applicable when the reference product would be classed as an NBCD, it is relevant to analyse the legislative provisions for these applications. As it is the case for chemical medicinal products, biosimilar applications are first expected to meet the conditions for the definition for generic medicinal products. It is only failing this definition, owing to, in particular, differences relating to raw materials or differences in manufacturing processes of the biological medicinal product and the reference biological medicinal product, that results of appropriate preclinical tests or clinical trials relating to these conditions must be provided. Reference is then made to the relevant criteria detailed in Annex I of the Directive. In this annex general requirements and reference to the relevant scientific guidelines are described. As for biosimilars, the Directive also directs the requirements for chemical generic and hybrid applications to the appropriate detailed guidelines (Article 10.2(b)). This is to say that with regards to legal provisions to dossier contents there is no significant difference to how simple or complex chemical products and biosimilars are addressed as for both general requirements and a reference to the applicable guidance is mentioned in the Directive.
Harmonization of requirements across regions
Follow-on products of nanomedicines, a representative group of NBCDs, have been named similar nanomedicines or nano similars [8], highlighting the challenges presented by these products due to a complexity closer to biosimilars than their chemical counterparts. An important aspect of biosimilars is the innovation brought by the ‘Guideline on similar biological medicinal products’ [9] to the origin of batches used in the in vivo comparability studies; the following is stated in that regard:
‘However, with the aim of facilitating the global development of biosimilars and to avoid unnecessary repetition of clinical trials, it may be possible for an Applicant to compare the biosimilar in certain clinical studies and in in vivo non-clinical studies (where needed) with a non-EEA authorised comparator (i.e. a non-EEA authorised version of the reference medicinal product) which will need to be authorised by a regulatory authority with similar scientific and regulatory standards as EMA (e.g. ICH countries). In addition, it will be the Applicant’s responsibility to demonstrate that the comparator authorised outside the EEA is representative of the reference product authorised in the EEA.’
As is the case for biosimilars, follow-on NBCDs may also require extensive in vivo comparability data. The flexibility accorded to biosimilars could be extended for NBCDs allowing for non-EEA reference medicinal products to be used, thus decreasing the need for duplicate studies of applicant wishing to apply for an abridged application in the EU and in another ICH country. Efforts in that direction are currently being made in the Transatlantic Trade and Investment Partnership [10] (TTIP) with the negotiations between the EU and US moving forward towards recognizing each other’s reference medicinal products. This is also an example of the narrowing of any existing differences of approach and flexibility between biosimilars and nanosimilars. The scope of TTIP also extends to the convergence of technical requirements between the EU and US – providing an additional drive to the harmonization to technical requirements to follow-on medicinal products.
Efforts to facilitate the worldwide harmonization of nanotechnology regulation are also demonstrated by the work of the International Pharmaceutical Regulators Forum (IPRF) with respect to a particular type of NBCDs, such as the nanomedicines. The purpose of the IPRF is for pharmaceutical regulators to exchange information on issues of mutual concern and regulatory cooperation. This exchange amongst global regulators will maximize potential efficiencies in addressing the increasingly complex global context of medicines regulation. IPRF intends to promote the implementation of ICH and other internationally harmonized pharmaceutical guidelines. It further contributes to the coordination of a range of international efforts related to regulation of medicines. The IPRF offers the opportunity to leverage expert scientific knowledge, regulatory and operational experience, ongoing technical harmonization work, and information access of other participating regulators. Goals include:
- Identifying new approaches and specific best practices, and developing smart strategies for dealing with the challenges of a rapidly evolving globalized pharmaceutical industry in the field of nanotechnology
- Providing a global overview of the different regulatory developments at national and international level and enable open sharing of information and ideas among regulatory leaders with hands-on operational responsibilities. This information sharing will allow the forum participants to discuss issues at an actionable level of detail
- Supporting international regulatory cooperation in areas that are not covered by existing initiatives
A special IPRF Working Group on Nanotechnology has been created in 2014 and its mandate and 2015/2016 work plan have been adopted and published in 2015 [11].
Deliverables for the IPRF 2015/2016 work plan include:
- Information sharing and mapping (annual regulatory updates)
- Compilation, mapping and discussion on terminology and definitions with focus on the classification of nanomedicines/nanotechnology in drug products
- Compilation of information for understanding synergies between the nano(medicine) safety and nano (medicine) toxicology fields
- Compilation of investigations required for ‘generic’ nanomedicines/nanotechnology in drug products used in regulatory procedures
- Mapping and exchange of requirements for nanomedicine/nanotechnology in drug product class specific guidance, e.g. liposomal formulations
- Exchange and mapping of general Critical Quality Attributes principles for nanomedicines/nanotechnology in drug products
In January 2016, the IPRF Nanotechnology Working Group published a document on ‘Information sharing and mapping’ highlighting the participating regulators’ current available guidance and status on related definitions, technical guidance in the respective region, planned workshops and trainings, legislation and classification system for related pharmaceuticals [12].
EMA scientific guidance
EMA has published a wide range of technical requirements to demonstrate the quality, safety and efficacy of medicinal products. These technical requirements documents are available as reflection papers and scientific guidance. The examples in Table 1 include the published reflection papers on nanomedicines and other guidance that may be applicable to NBCDs; for a full list of scientific guidance published please go to the EMA’s guidance webpage [13].

The provision of scientific advice by National Competent Authorities or EMA [14] is available to applicants. Scientific advice on the appropriate tests and studies in the development of a medicine is designed to facilitate the development and availability of high quality, effective and acceptably safe medicines, for the benefit of patients. During the development of most nanomedicines scientific advice was requested from EMA to enhance the chances for a positive development outcome.
Developers of novel medical products are also encouraged at an early stage of development to seek advice of the EMA’s Innovation Task Force (ITF) [15].
The initiatives described above for harmonization across regions and published guidance, together with the provision of Scientific Advice and ITF meetings, are examples of how EMA aims at facilitating the development of innovative medicines.
Table 2 provides examples of products approved in the EU, through centralized and national authorization procedures, that could be categorized as NBCDs [16].
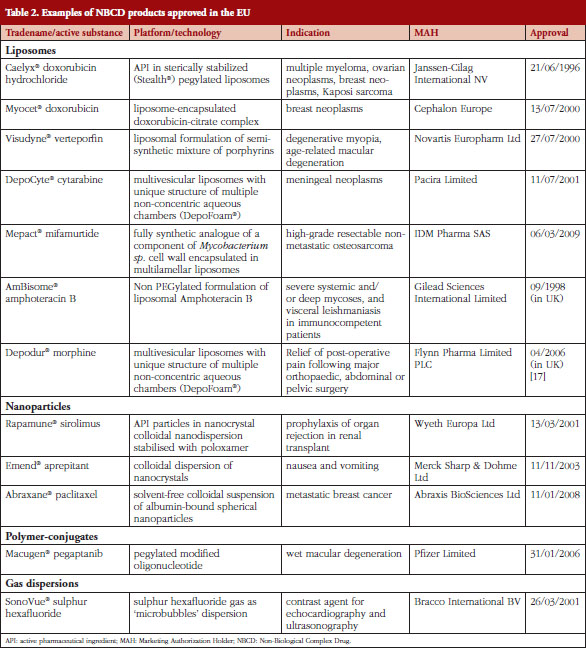
It is noteworthy that first-generation nanomedicines, including liposomal formulations, iron-based preparations and drug nanocrystal technologies in oral dosage forms, have been established as safe and effective for many years. EMA has evaluated marketing authorization applications for nanomedicines thus demonstrating the suitability of the current legislation and scientific guidance to accept complex medicinal products; the basis on which the scientific opinions were made on the marketing authorization submitted for NBCDs are publicly available [18]. Orphan status has been granted for ten nanomedicines on recognition by EU regulators of the potential benefits stemming from the use of nanotechnology.
Recent publications confirm that approximately 48 nanomedicines and nanoimaging agents are currently under clinical development (phase I–III) in Europe, with others progressing through earlier stages of drug discovery and non-clinical development [19]. In addition, approximately 70 cancer clinical trials are ongoing in the US involving nanomedicines and, therefore, the number of marketed pharmaceuticals using nanotechnology is expected to continuously grow and, thus, benefit patients and public health.
In conclusion, the current EU regulatory framework for medicinal products has been proven to be robust and efficient for the evaluation of NBCDs, including nanomedicines, facilitating the deliverable of safe and efficacious medicines to patients. The EU network recognizes the potential benefits deriving from innovative technologies. Efforts are being undertaken to continuously develop and harmonize across regions the regulatory requirements for complex medicinal products in view of promoting timely access of safe and effective medicines to patients. The EU network is ready for NBCDs.
Competing interests: The views expressed are those of the authors and should not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency.
Provenance and peer review: Commissioned, externally peer reviewed.
Authors
Falk Ehmann, MD, PhD, MSc
Clinical Pharmacology – Science and Innovation
European Medicines Agency
30 Churchill Place, Canary Wharf
London E14 5EU, UK
Ruben Pita, PharmD, LLM
Procedure Management Department
European Medicines Agency
30 Churchill Place, Canary Wharf
London E14 5EU, UK
References
1. Schellekens H, Stegemann S, Weinstein V, de Vlieger JS, Flühmann B, Mühlebach S, et al. How to regulate nonbiological complex drugs (NBCD) and their follow-on versions: points to consider. AAPS J. 2014;16(1):15-21.
2. Flühmann B, Crommelin DJA, de Vlieger JSB, Borchard G, Scott E McNeil, Mühlebach S, et al. Non-Biological Complex Drugs (NBCDs) and their follow-on versions: time for an editorial section. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):167-70. doi:10.5639/gabij.2015.0404.037
3. European Commission. EU Pharmaceutical Informations. EudralLex Volume 1 – Pharmaceutical legislation medicinal products for human use [homepage on the Internet]. [cited 2016 Apr 1]. Available from: http://ec.europa.eu/health/documents/eudralex/vol-1/index_en.htm
4. Heads of Medicines Agencies. CMDH What’s new [homepage on the Internet]. [cited 2016 Apr 1]. Available from: http://www.hma.eu/cmdh.html
5. European Medicines Agency. Reflection paper on the data requirements for intravenous liposomal products developed with reference to an innovator liposomal. EMA/CHMP/806058/2009/Rev. 02. 21 February 2013 [homepage of Internet]. [cited 2016 Apr 1]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500140351.pdf
6. European Medicines Agency. Guideline on the investigation of bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1/Corr**. 20 January 2010 [homepage of Internet]. [cited 2016 Apr 1]. Available from: http://www.ema.europa.eu/ema/pages/includes/document/open_document.jsp?webContentId=WC500070039
7. Borchard G, Flühmann B, Mühlebach S. Nanoparticle iron medicinal products – requirements for approval of intended copies of non-biological complex drugs (NBCD) and the importance of clinical comparative studies. Regul Toxicol Pharmacol. 2012;64(2):324-8.
8. Ehmann F, Sakai-Kato K, Duncan R, Pérez de la Ossa DH, Pita R, et al. Next-generation nanomedicines and nanosimilars: EU regulators’ initiatives relating to the development and evaluation of nanomedicines. Nanomedicine (Lond). 2013;8(5):849-56.
9. European Medicines Agency. Guideline on similar biological medicinal products. CHMP/437/04 Rev 1. 23 October 2014 [homepage on the Internet]. [cited 2016 Apr 1]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf
10. European Commission. Technical paper for regulatory cooperation on generic medicines. Proposal of the European Union. 26 January 2016 [homepage on the Internet]. [cited 2016 Apr 1]. Available from: http://trade.ec.europa.eu/doclib/docs/2016/january/tradoc_154172.pdf
11. International Pharmaceutical Regulators Forum. IPRF Nanomedicines Working Group [homepage on the Internet]. [cited 2016 Apr 1]. Available from: https://www.i-p-r-f.org/en/working-groups/nanomedicines-working-group/
12. International Pharmaceutical Regulators Forum. IPRF Nanomedicines Working group – Information sharing and mapping (as of January 2016) [homepage on the Internet]. [cited 2016 Apr 1]. Available from: https://www.i-p-r-f.org/files/7414/5387/7778/IPRF_Nanomedicines_WG_-_info_mapping_Jan2016.pdf
13. European Medicines Agency. Scientific guidelines [homepage on the Internet]. [cited 2016 Apr 1]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000043.jsp&mid=WC0b01ac05800240cb
14. European Medicines Agency. Scientific advice and protocol assistance [homepage on the Internet]. [cited 2016 Apr 1]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000049.jsp&mid=WC0b01ac05800229b9
15. European Medicines Agency. Innovation Task Force [homepage on the Internet]. [cited 2016 Apr 1]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000334.jsp&mid=WC0b01ac05800ba1d9
16. Adapted from Crommelin, Daan JA, and Jon SB de Vlieger, eds. Non-Biological Complex Drugs: The Science and the Regulatory Landscape. Vol. 20. Springer, 2015.
17. Flynn Pharma Ltd. Depodur marketing ceased [homepage on the Internet]. [cited 2016 Apr 1]. Available from: http://www.flynnpharma.com/about-us/news/depodur-marketing-ceased
18. European Medicines Agency. European public assessment reports [homepage on the Internet]. [cited 2016 Apr 1]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&;mid=WC0b01ac058001d124
19. Noorlander CW, Kooi MW, Oomen AG, Park MV, Vandebriel RJ, Geertsma RE. Horizon scan of nanomedicinal products. Nanomedicine (Lond). 2015;10(10):1599-1608.
*A biological medicinal product is a product, the active substance of which is a biological substance. A biological substance is a substance that is produced by or extracted from a biological source and that needs for its characterization and the determination of its quality a combination of physicochemical-biological testing, together with the production process and its control – definition from Directive 2001/83/EC.
|
Author for correspondence: Falk Ehmann, MD, PhD, MSc, Clinical Pharmacology – Science and Innovation, European Medicines Agency, 30 Churchill Place, Canary Wharf, London E14 5EU, UK |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


