The EU regulatory network and emerging trends – a review of quality, safety and clinical development programmes
Published on 2021/02/23
Generics and Biosimilars Initiative Journal (GaBI Journal). 2021;10(2):83-99.
Author byline as per print journal:
Marta Zuccarelli1, PharmD; Benjamin Micallef1, PharmD; Mark Cilia1, PharmD; Anthony Serracino-Inglott1,2, PharmD; John-Joseph Borg1,3, PhD
|
Introduction/Study Objectives: The development of biosimilars is challenging due to the complexity of the active substances as well as the strict regulatory requirements to show similarity with a reference medicinal product. This review aims to describe the regulatory experience of approving biosimilars in the European Union (EU) within the EU framework, identify emerging trends in the EU regulatory pathway when approving biosimilars and discuss where the EU biosimilar framework is heading. |
Submitted: 3 December 2020; Revised: 11 February 2021;Accepted: 15 February 2021; Published online first: 1 March 2021
Introduction/Study Objectives
The regulatory framework in the EU
According to the European Union (EU) law, a similar biological medicinal product or ‘biosimilar’ is a biological medicinal product which is similar, but not identical, to a reference biological medicinal product [1]. Biosimilar products in the EU would have demonstrated similarity to the reference medicinal product in terms of physicochemical characteristics as well as safety and efficacy through a comprehensive comparability exercise [2].
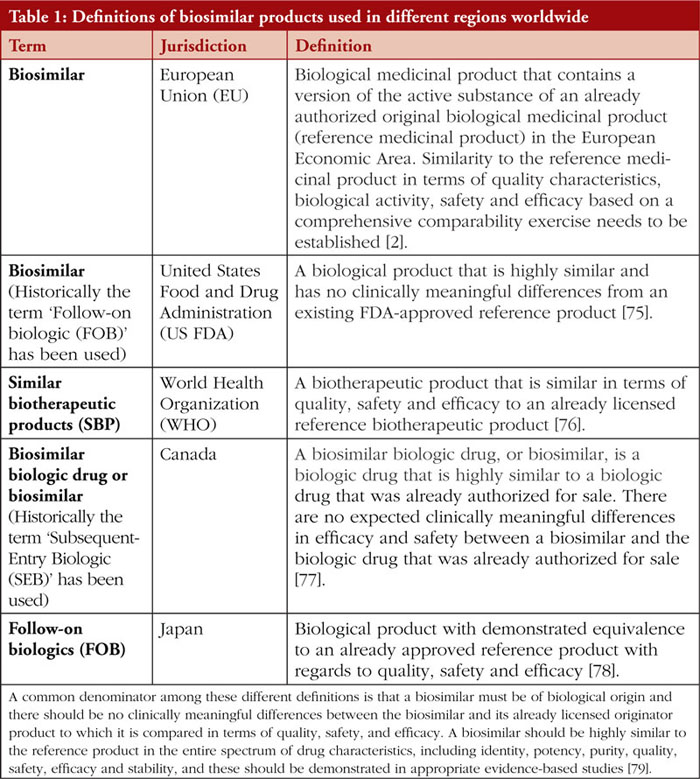
It is important to note that different regions worldwide have varying definitions of biosimilar products or equivalent terms, see Table 1, and have different requirements surrounding biosimilar clinical interchangeability, switching and substitution [3].
In order to understand the complexities of the EU regulatory framework surrounding the approval of biosimilars, it is important to clarify that the approval of a biosimilar in the EU is based on an ‘abridged’ marketing authorisation application (MAA), where similar biological products do not require full scientific data as for new biological applications [4]. In principle, biosimilars can be developed for any well characterized biological product [5] which is produced by biotechnological methods, however, biosimilars are unlikely to be produced for vaccines, allergens, blood or plasma-derived products and gene/cell therapy products.
In the EU, the regulatory framework on biosimilars is built on guidelines, with the European Medicines Agency (EMA) issuing the first guideline on similar biological medicinal products in 2005 [5]. This overarching guideline provided complementary directions to the legal basis set out by the Directive 2001/83/EC in 2004 [1]. Since 2005, an additional two overarching guidelines dealing with non-clinical and clinical issues and on quality were also published [6]. Furthermore, product-specific guidelines laying down scientific recommendations for the development of biosimilars were issued for similar biological medicinal products containing monoclonal antibodies, recombinant follicle stimulation hormone, recombinant interferon beta, recombinant erythropoietin, low-molecular-weight heparins (LMWHs), recombinant interferon-alfa, recombinant granulocyte colony stimulating-factor (G-CSF), somatropin and recombinant human insulin [7], see Figure 1.

As a consequence of the regulatory framework set up in the EU since 2005, there has been a steady increase in the number of biosimilar medicinal products during the last 20 years, with successful use in treating and managing many chronic, debilitating and life-threatening diseases, such as autoimmune diseases and various forms of cancer [8].
Biosimilarity is proven through the use of a stepwise approach, first in quality, then in the non-clinical setting [using comparability exercises in pharmacokinetics (PK) and pharmacodynamics (PD) both in vitro and in vivo and then clinically [9].
In this study we aimed to describe and discuss the regulatory experience of approving biosimilars in the EU (from 2005–2019), through qualitative and quantitative evaluations. In addition, we discuss how the EU regulatory network has fared and what are the noteworthy emerging trends in biosimilar development as identified through a review of quality, safety and clinical development programmes. The results of this review are of interest to both developers and regulators and may help in understanding the changing landscape of biosimilar regulation.
Methods
Review of clinical development programmes
The number of MAAs for biosimilar medicinal products submitted for regulatory review up to December 2019 was retrieved from the EMA website [10]. The applications submitted for biosimilars were grouped as MAAs currently under review and MAAs which reached the post-authorization phase. The latter group was further subdivided in line with the opinion given to each biosimilar (either positive or negative) and MAAs which were withdrawn. Biosimilar medicinal products which received a positive opinion and maintained a valid authorization were further analysed. To detect emerging trends, a comparison of clinical development programmes of biosimilar products with comparable clinical development was carried out.
Using the EMA database of European public assessment reports (EPARs), biological originators and the number of biosimilars for each originator were retrieved. The EPARs of biosimilar medicinal products with a valid marketing authorization (up to December 2019) were used as the main source of information [11-58].
Information on clinical development programmes of each biosimilar were compared with the approved guidelines [7] to detect any notable considerations and consequent justification. The extrapolation of indication was analysed for applicable biosimilar medicinal products.
It should be noted that not all biosimilar medicinal products analysed were different products. Some companies conducted one clinical development programme for the biosimilar and then marketed the same product under different names. Biosimilar products having the exact same clinical development programme, but different trade names are indicated with a slash, e.g. Inflectra®/Remsima®, in the results section.
Safety
Since the safety of biosimilar medicinal products could be of a ‘potential’ concern, static reporting odds ratio (ROR) reports [at the System Organ Class (SOC) level] concerning each biosimilar medicinal product were retrieved from the EudraVigilance data analysis system (EVDAS). SOCs ‘product issues’ and ‘social circumstances’ were excluded a priori because they were not considered relevant for the purpose of this review. To detect any new disproportionate reporting that may have arisen in the post-marketing phase, adverse event data were captured separately for the period before the authorization date of the first biosimilar reaching the market and period after authorization (up to a cut-off date 25 June 2020). Adverse event data from the two periods was then compared.
The expectedness of new disproportionate reporting identified from period after the first biosimilar authorization was evaluated in line with the summary of medicinal product characteristics (SmPCs) of the reference product. For each new disproportionate SOC reaction, the following SmPC sections were evaluated; 4.4 – Special warnings and precautions for use, 4.6 – Fertility, pregnancy and lactation, and 4.8 – Undesirable effects.
For SOCs which were deemed unexpected, i.e. not listed in the above-mentioned sections of the SmPCs, another static ROR evaluation was performed to identify which preferred terms (PTs) were disproportionately reported within each SOC. PTs which were disproportionately reported were evaluated for expectedness in line with the SmPC of the reference product (as described previously). Disproportionate PTs which were unexpected were then compared to EMA’s designated medical event (DME) list [59]. The PT, ‘fetus malformation’ was also considered (in addition to DMEs) as a serious adverse event of interest for the analysis we carried out. Disproportionate PTs which were unexpected, and which were also DMEs were further assessed by performing a causality assessment using the French imputability method [60] to confirm or confute potential signals of disproportionate reporting.
Results
The experience gained reviewing biosimilar marketing authorisation applications
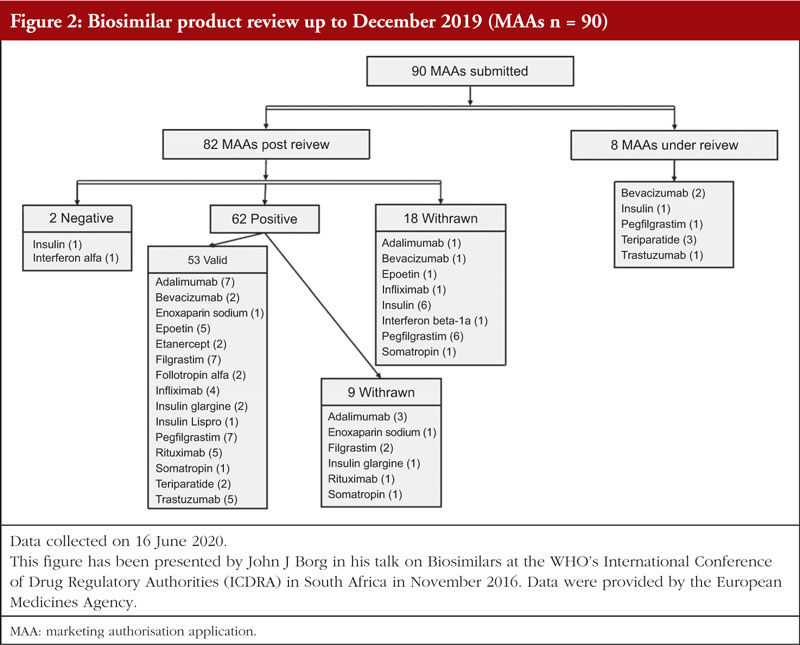
Since 2004, EMA has gained significant experience in the scientific evaluation of biosimilar applications and revision of the regulatory scientific guidelines for the European markets was observed, see Figure 1.
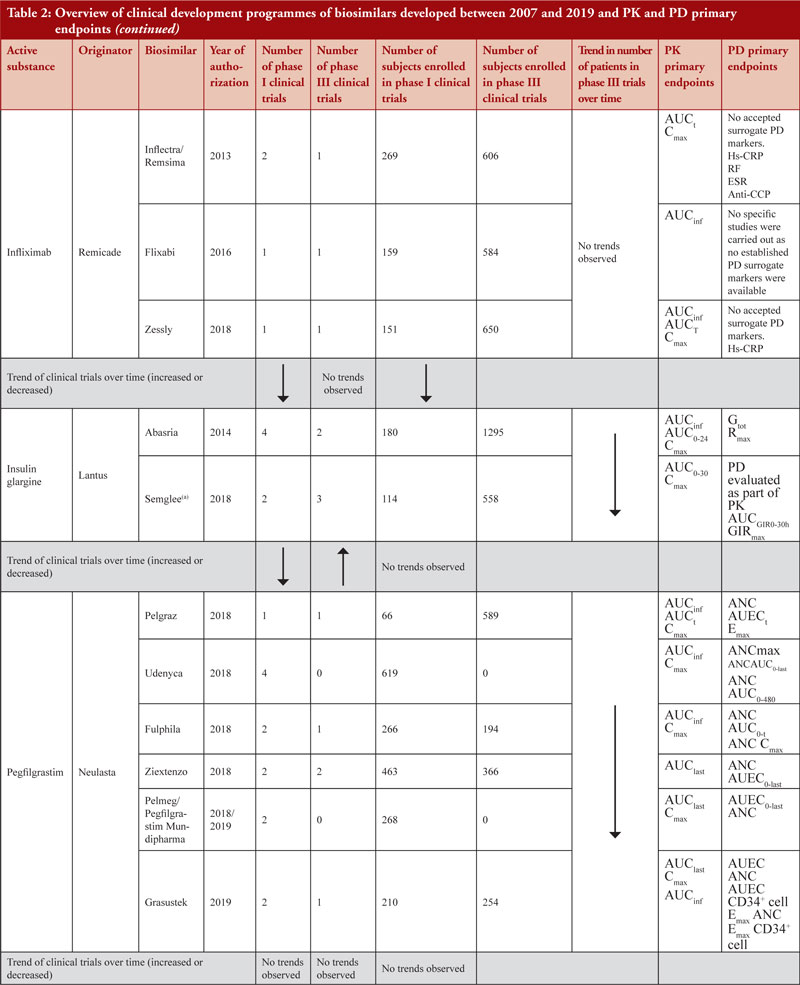
Up to December 2019, 90 MAAs had been submitted to EMA for review. Figure 2 shows the authorization status of each MAA as well as applications under review. Fifty-three biosimilar products held valid marketing authorization in Europe as of December 2019. These 53 biosimilars resulted from 35 unique development programmes and were based on 14 different reference biologicals (adalimumab, bevacizumab, enoxaparin sodium, epoetin, etanercept, filgrastim, follitropin alfa, infliximab, insulin, pegfilgrastim, rituximab, somatropin, teriparatide, trastuzumab).
Biosimilar guidelines: evolution in EU – totality of evidence founded on quality data
In Europe, guidelines were issued for biosimilars to help applicants prepare MAAs. The first guidelines for similar biological medicinal products was published in October 2005. Since 2005, an additional two overarching guidelines (one on non-clinical/clinical issues and one on quality), eight product-specific guidelines and one reflection paper were published, see Figure 1. The reflection paper will be addressed as a product-specific guideline throughout this review. The first product-specific guideline was published in 2006 to discuss non-clinical and clinical studies recommended during the development of insulin’s biosimilars. Following the first guidelines on insulin, product-specific guidelines on somatropin, recombinant G-CSF, epoetin, interferon alfa (IFN-α), LMWH, monoclonal antibodies (mAbs), interferon beta (IFN-β) and recombinant follicle-stimulating hormone (follitropin) were published between 2006 and 2012. Six out of nine product-specific guidelines (insulin, somatropin, G-CSF, epoetin, LMWH, follitropin) were revised between 2015 and 2019, see Figure 1. It was observed that for certain biosimilar classes, revision of guidelines was related to more experience and knowledge gained by regulators over time. For example, the first guidelines on G-CSF emphasized the performance of ‘at least one repeat dose toxicity study in a relevant species’ and the performance of ‘confirmatory clinical trials to compare efficacy and safety of the biosimilar and reference’. In the revised draft of the guidelines for G-CSF, in vivo studies are no longer recommended, and confirmatory efficacy clinical trials are not deemed necessary as ‘pivotal evidence for similar efficacy will be derived from the similarity demonstrated in physicochemical, fu-nctional, pharmacokinetic and phar-macodynamic comparisons’ [61-62]. Another example of guideline revision is the LMWH guideline. In the first issue of the LMWH guidelines, in vivo PD studies were always deemed necessary, but in the revised guidelines, in vivo PD studies are not required as part of the comparable studies unless physicochemical and biological comparability has not been already demonstrated. According to the revised LMWH guidelines, toxicological studies are only required in specific cases, while in the first issue of the same guideline, at least one repeated dose toxicity study in one relevant species was requested. In addition, according to the revised LMWH guidelines, clinical efficacy studies are not required any longer as efficacy data will be ‘derived from the similarity demonstrated in physicochemical, functional and pharmacodynamic comparisons’ [63-64].
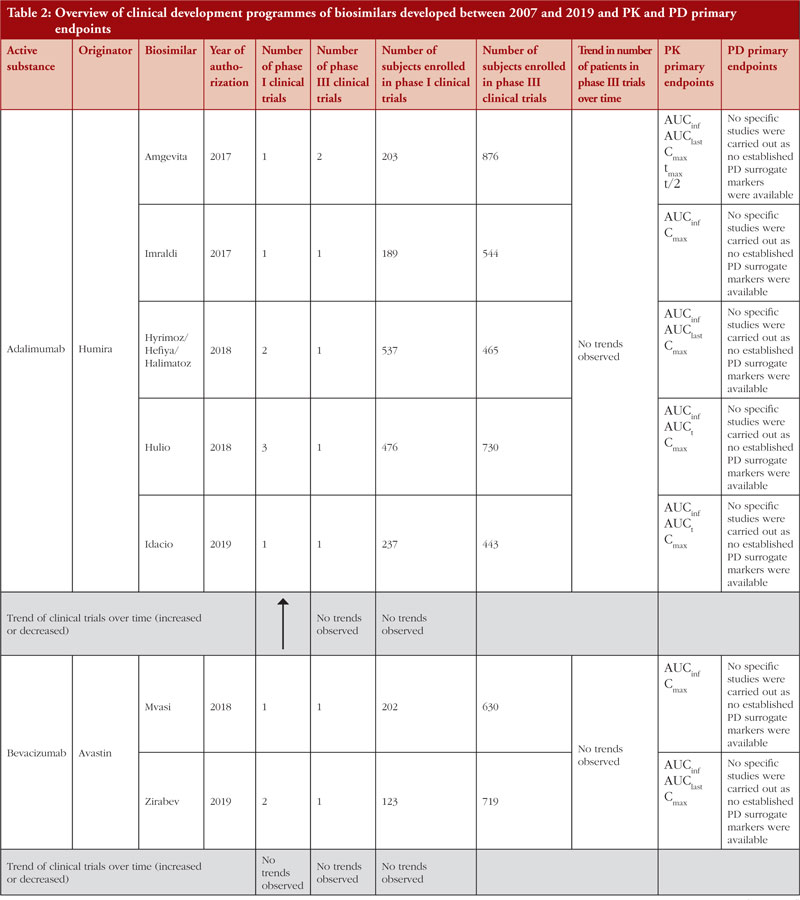
The biosimilar guidelines are not meant to be prescriptive step-by-step guides on how to design a clinical study programme to obtain approvals for biosimilars but rather these guidelines contain scientific recommendations to facilitate development of biosimilars through comparability programmes with a reference medicinal product. The biosimilar comparability exercise is pivotal to understanding the degree of similarity between test and reference products and is crucial in the benefit-risk assessment of biosimilar products. Initially, biosimilar guidelines could have been considered as conservative when laying down clinical requirements required by a manufacturer to prove biosimilarity. Where, for example, in the case of epoetins: two clinical studies are required in titration and maintenance phase with additional emphasis on animal data to be provided before licensure. Also, the number of patients, number of studies and duration of studies are three important parameters that should be taken into consideration whilst designing a clinical development programme as these parameters determine the success and efficiency of a clinical developmental programme. Notwithstanding these considerations, it is interesting to note that revised product-specific guidelines have re-considered the regulatory requirements on clinical data and in general, have shifted to rely more on extrapolation with usage of PD markers for clinical endpoints as a surrogate for efficacy and additional supportive evidence from relevant non-clinical in vivo studies. The strategy behind this shift is to have an increased reliance on the detailed and comprehensive quality data required in the marketing authorization dossier. This shift in regulatory requirements is based on the regulatory experience obtained to date approving biosimilars by EU regulators. In order to better understand the experience we have to date in approving biosimilars, we reviewed the clinical development programmes of 11 biosimilar active substances (adalimumab, bevacizumab, epoetin, etanercept, filgrastim, follitropin alfa, infliximab, insulin glargine, pegfilgrastim, rituximab, trastuzumab) to identify any emerging common trends between classes as described in their EPARs. A comparison was also made between biosimilars clinical development programmes of the same class.
Clinical development programmes of biosimilars
Clinical development programmes of 48 out of the 53 currently authorized biosimilar medicinal products were compared. Enoxaparin sodium, insulin lispro and somatropin were not included because only one biosimilar medicinal product was developed or maintained a valid authorization at the time of review, therefore a comparison could not be performed. Teriparatide was not included in the comparison exercise since the two biosimilars authorized were based on the same development programme and were then sold under different brand names.
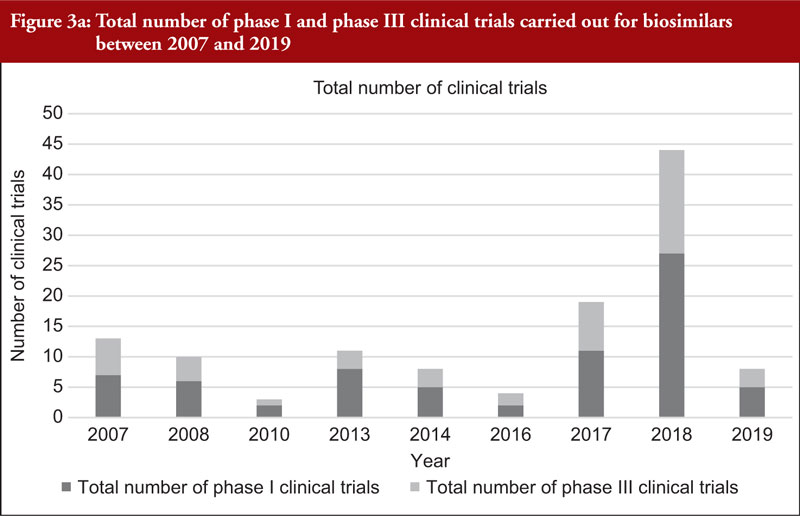
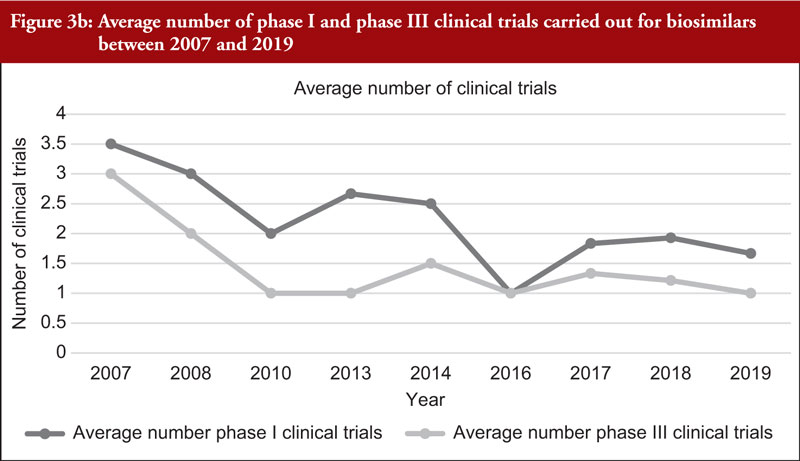
When considering the 48 biosimilar medicinal products compared, it was observed that, in general, total number of clinical trials (both phase I and phase III) across all biosimilars increased over time, see Figure 3a. This is not unexpected since the total number of biosimilar approvals increased over time, with the number of approvals peaking in 2018 when 16 biosimilars were approved based on 14 clinical development programmes, see Table 2. The number of studies leading to approval changed over time. In 2007, the average number of phase I clinical trials was 3.5 and the average number of phase III clinical trials was 3. In 2019, the average number of phase I clinical trials was 1.67 and the average number of phase III clinical trials was 1, see Figure 3b, with certain biosimilars of pegfilgrastim, such as Pelmeg®/Pegfilgrastim Mundipharma® and Udenyca®, not having carried out any phase III clinical trials.
Enrolled subjects
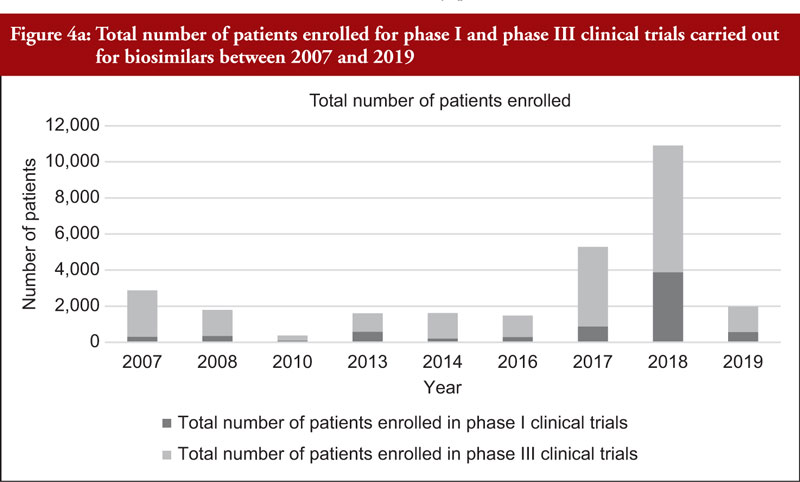
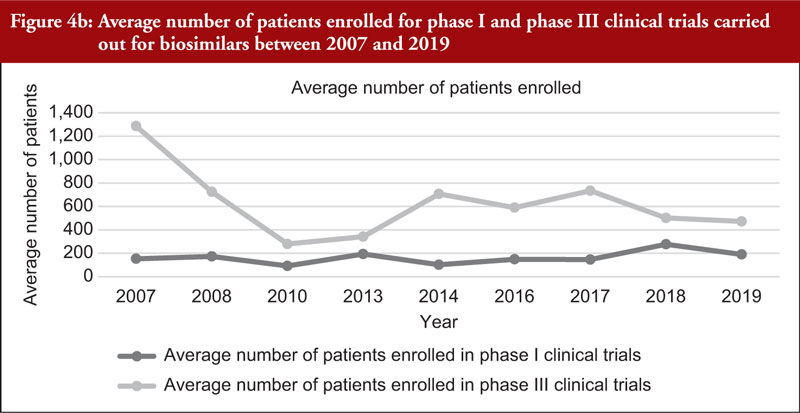
The total number of subjects enrolled in clinical trials (phase I and phase III combined) across all biosimilars compared was observed to have generally increased through time although a high variability in the number of enrolled subjects per development programme was noted. We also evaluated the number of subjects enrolled in phase I and phase III clinical trials (not combined), see Figure 4a. Figure 4a shows an increased total number of subjects enrolled in phase I and phase III clinical trials through time although the increased number of biosimilar medicinal products authorized in more recent years should be considered. For this reason, we evaluated the average number of subjects enrolled in clinical trials as shown in Figure 4b. From Figure 4b, it can be appreciated that the average number of subjects enrolled in phase I clinical trials was mostly stable. In phase III clinical trials, it can be observed that the average number of subjects enrolled steadily increased between 2010 and 2017 and decreased from 2017 to 2019.
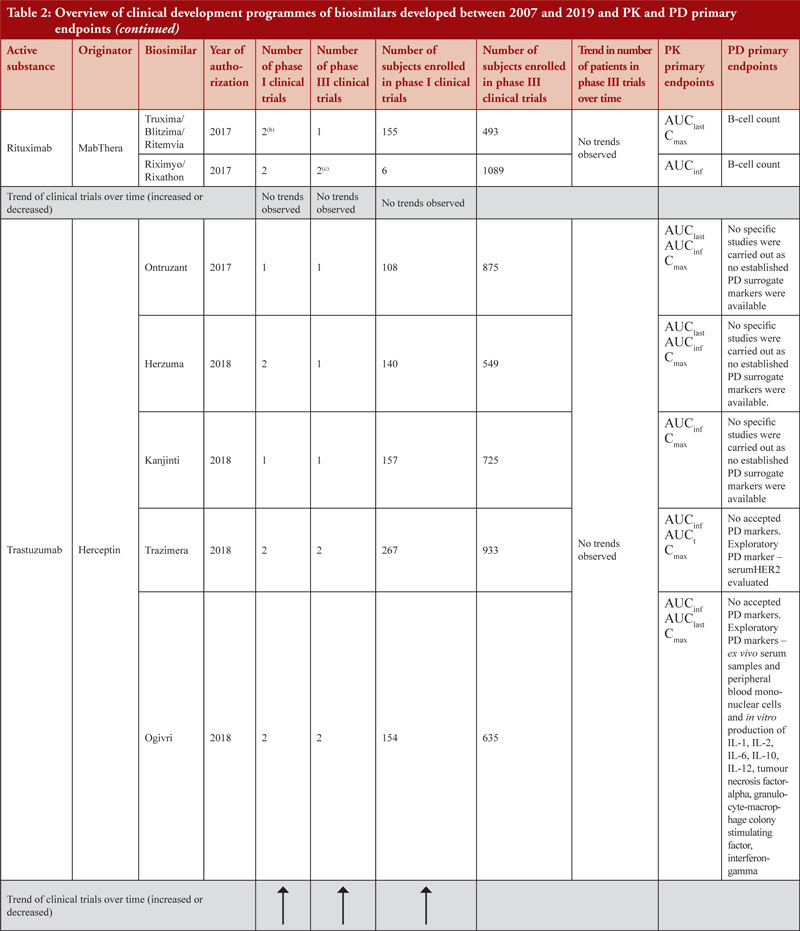
Healthy volunteers were enrolled for phase I clinical trials for all biosimilars clinical development programmes, except for insulin glargine biosimilars (where both healthy volunteers and patients with type 1 diabetes mellitus were enrolled), infliximab biosimilars (where only patients with rheumatoid arthritis (RA) were enrolled and in the case of Inflectra®/Remsima® in addition to RA patients, patients with ankylosing spondylitis were also enrolled) and rituximab biosimilars (where only patients with RA and diffuse large B-cell lymphomas were enrolled).

Primary endpoints to support pivotal data
Demonstration of pharmacokinetic (PK) equivalence between the test product and reference product is deemed necessary for the approval of a biosimilar. PK was observed to be mainly studied in phase I clinical trials on healthy volunteers since a cohort of healthy volunteers is considered to be a more heterogeneous population. Supportive PK data are derived from phase III clinical trials. During the clinical development programmes of insulin glargine biosimilars, infliximab biosimilars and rituximab biosimilars, PK studies were conducted on patients where, AUCinf, AUClast and Cmax with an acceptance level of between 0.8 and 1.25 with 90% confidence interval (CI) were the primary PK endpoints used to evaluate bioequivalence between test product and reference product, see Table 2.
There have been examples where therapeutic equivalence was demonstrated despite PK study results not falling within the pre-specified acceptance level. During the development of adalimumab biosimilars Hyrimoz®/Hefiya®/Halimatoz®, a second phase I PK clinical trial (study GP17-104) was deemed necessary after two out of three primary PK endpoints (AUCinf and AUC0last) were above pre-specified acceptance level in the first phase I PK clinical trial (study GP17-101) carried out. In the second PK study, the study design was adapted, and the primary endpoints were reduced to two (with AUCinf, Cmax and AUClast not calculated). The results obtained from the second PK study were within the pre-specified acceptance level of 0.8–1.25 with 90% CI. In this case, PK was further investigated in a phase III clinical trial (study GP17-301) before biosimilarity between the test product and the reference product could be definitively confirmed [13-15].
It is observed that, in some cases, the acceptance level of PK parameters was changed in post-hoc analysis and such an approach was considered acceptable when considering the justification provided. For epoetin zeta biosimilars, the primary endpoints were evaluated with 90% CI and the acceptance level for Cmax was widened by the applicant to 0.7%–1.43% in the post-hoc analysis. In the EPAR, it is explained that the applicant ‘referred to the Scientific Advice given by CHMP in April 2004 that stated that the concept of “comparability” cannot use bioequivalence but that similar PK profiles of SB309 and the reference product would strengthen the choice of reference in the clinical trials. The advice concluded that for this purpose descriptive statistics will suffice’ [23, 24].
PD studies to evaluate clinical comparability between test product and reference product are recommended by the guidelines, however, for active substances adalimumab, bevacizumab, epoetin, etanercept, infliximab and trastuzumab, no established PD surrogate markers were available at the time when the studies were conducted. Due to lack of accepted PD markers there were cases in which no PD studies were carried out. Clinical comparative PD studies were not conducted for any of the adalimumab biosimilars as no specific and accepted PD markers to predict efficacy exists. For all adalimumab biosimilars except for Hulio®, the EPAR states that only in vitro PD studies were carried out during the development of the products. For Hulio®, the mechanism of action (MOA) is described, but no PD studies are mentioned [16].
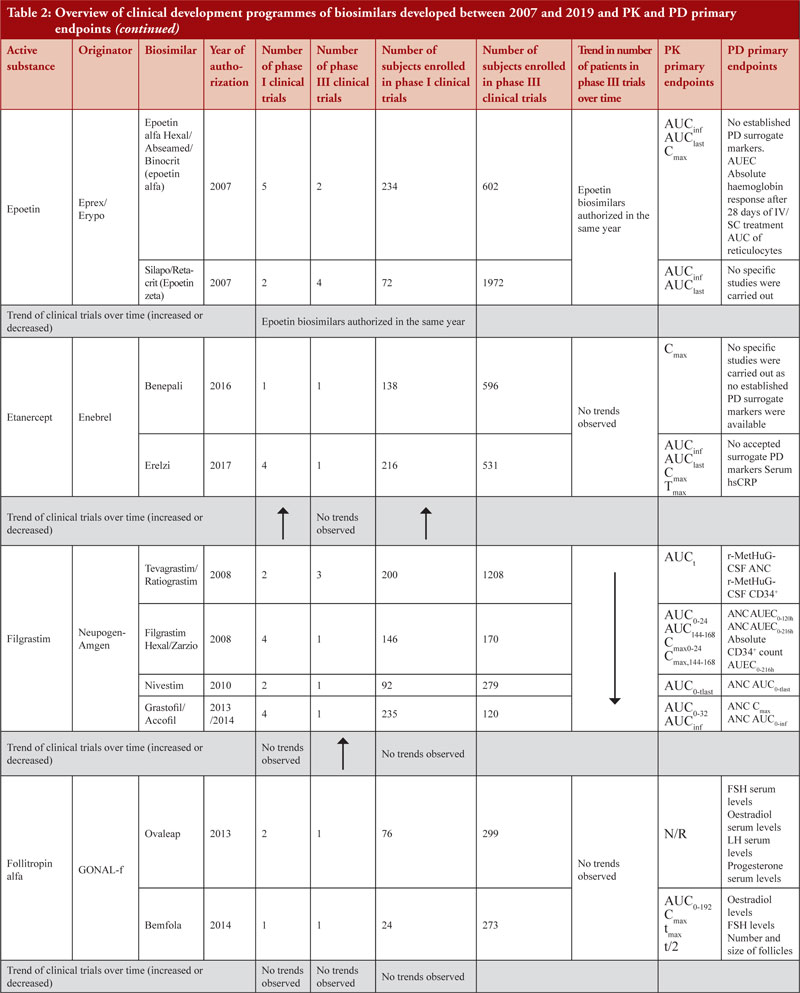
PD surrogate markers were explored by applicants for certain products. For example, during the clinical development programme of epoetin alfa biosimilars (epoetin alfa hexal®/Abseamed®/Binocrit®), PD studies were conducted as part of PK studies with PD markers set by the applicant [20-22]. However, during the clinical development programmes of epoetin zeta biosimilars, no studies were conducted to define the PD profile.
During the clinical development programme of Erelzi®, PD was indirectly evaluated in a phase III clinical trial. The PD marker serum high-sensitivity C-reactive protein (hs-CRP) was measured, showing a similarity between Enbrel® and Erelzi®. However, in the EPAR it is stated that data on serum hs-CRP should be considered as supportive since this is not a specific biomarker [26]. It is interesting to notice that there are no specific biomarkers available for tumour necrosis factor-alpha (TNF-α) inhibitor and that clinical comparability can be demonstrated also by clinical evidence other than PD surrogate [65, 66].
Another example for which no PD markers are accepted surrogates is represented by infliximab biosimilars although the applicant explored CRP for Inflectra®/Remsima® and Zessly®. In the Zessly®’s EPAR it is stated that ‘CRP is a marker of disease activity but does not have a clear relationship to therapeutic effect’ [39]. By comparing CRP differences between the reference products and the test product, similarity was evaluated. For well-known active substances, such as filgrastim, more stringent PD criteria were required by regulators, as observed for both Filgrastim Hexal® and Zarzio®, where the PD bioequivalence criteria (80% to 125%) at 95% CI was not accepted as an acceptance criterion for PD studies, as stricter acceptance criteria were expected (+/- 10%). The rationale for a stricter acceptance criterion is explained in the EPAR as follows, ‘The predefined equivalence boundaries were derived by the applicant from published data on the effect observed for Neupogen compared to placebo. It was assumed that the smallest clinically relevant difference in PD response between the test and reference product was 15% of the effect observed for Neupogen® compared to placebo in the published study. Decreasing this margin to 10%, which approximately corresponds to half the increase in the area under the effect curve (AUEC) between the 2.5 µg/kg and 5 µg/kg doses, would result in more acceptable equivalence intervals; indeed, the 95% CI for ANC AUEC and Emax in study EP06-103 would still fall within these tighter equivalence boundaries’ [29, 30].
Data also show that for highly characterized active substances, such as in the case of pegfilgrastim biosimilars Udenyca®, Pelmeg® and Pegfilgrastim Mundipharma®, no phase III clinical trials were required, see Table 2.
Extrapolation of indication
According to the CHMP, safety and efficacy data can be extrapolated from studies of a biosimilar for a different indication than the one studied during a clinical trial. Extrapolation of safety and efficacy data is based on physicochemical, functional, non-clinical and clinical data available showing similarity between the biosimilar and the reference product. A dedicated section on extrapolation of data is present in eight out of nine product-specific guidelines (insulin, somatropin, G-CSF, epoetin, IFN-α, LMWH, mAbs, IFN- β and recombinant follicle-stimulating hormone).
During the review of the clinical development programmes, it has been observed that extrapolation of data has been a common approach when approving biosimilars that usually have the same therapeutic indications as the originator in spite of the fact that the biosimilar may not have been studied in any indication or has been studied in only one indication.
For example, biosimilars of infliximab have the same indications as the infliximab originator Remicade® even though biosimilars of infliximab have only been tested in patients with RA (and in the case of Inflectra®/Remsima® also in ankylosis spondylitis), while originator Remicade® was tested in patients with rheumatoid arthritis, Crohn’s disease, ulcerative colitis, ankylosis spondylitis and psoriatic arthritis. To allow for extrapolation of data from the clinical development programme of Remicade®, the biosimilarity of Inflectra®/Remsima® was demonstrated through a comparability exercise as is detailed in the product-specific guidelines. Based on the robust comparisons of the physicochemical and in vitro and ex vivo biological analyses, and together with clinical data demonstrating pharmacokinetic and therapeutic equivalence in rheumatology conditions, Inflectra®/Remsima® was considered biosimilar to the reference product Remicade and extrapolation to all other indications of Remicade was considered appropriate [36, 37]. The choice of using patients with RA during the development of Inflectra®/Remsima® to prove clinical efficacy is due to the more sensitive endpoints used in RA compared to endpoints used in Crohn’s disease and ulcerative colitis.
Extrapolation of indication does not always require comparability of efficacy data. In the case of clinical development programmes of Udenyca® and Pelmeg®/Pegfilgrastim Mundipharma® no phase III clinical trials on affected patient populations were carried out. Only clinical trials on healthy volunteers were submitted and therefore no comparability efficacy data were generated. Extrapolation of the indication was performed based on PK/PD similarity data, immunogenicity and safety. The absolute neutrophil count (ANC), a well-established PD marker, was used to established PD comparability. This is in line with the EMA biosimilar guidelines on recombinant granulocyte-colony stimulating factor (EMEA/CHMP/BMWP/31329/2005), according to which extrapolation of indication can be done if PD similarity with the reference product is demonstrated on healthy volunteers using ANC as a marker [61, 62].
In certain cases, the extrapolation of indication is restricted to certain specific indications. In the application of Silapo®/Retacrit®, the indication ‘reduction of allogeneic blood transfusions in adult non-iron deficient patients prior to major elective orthopaedic surgery’ was not granted because of lack of safety and efficacy data for the subcutaneous (SC) route of administration. The EPARs of Epoetin Alfa Hexal®/Abseamed®/Binocrit® state that ‘the claimed indications initially included also reduction of allogeneic blood transfusions in adult non-iron deficient patients prior to major elective orthopaedic surgery. However, during the CHMP scientific assessment it became evident that ‘efficacy and safety of Epoetin zeta have not been demonstrated for the SC route of administration in immunocompetent patients’ as well as ‘The benefit-risk ratio is not considered positive for the major orthopaedic surgery indication because immunogenicity of SC administered SB309 has not been assessed in immunocompetent individuals’ [20-22].
Extrapolation of safety and efficacy data is addressed by regulators in a specific section within the EPAR. For example, in the EPAR of Biograstim® (approved in 2008; biosimilar of Neupogen® (filgrastim)), information on extrapolation of safety and efficacy data is reported in the EPAR section ‘Overall conclusions, risk/benefit assessment and recommendation’ within the ‘Risk-benefit assessment’ subsection. The extrapolation of data was accepted with uncertainties and the outstanding issue was then tackled post-authorization in the products’ life cycle as an issue within the risk management plan. In the EPAR of Accofil®, a biosimilar containing filgrastim approved in 2014, extrapolation is discussed in the benefit-risk balance section, in the ‘Uncertainty in the knowledge about the beneficial effects’ subsection. In the EPARs of Biograstim® and Accofil®, uncertainties on biosimilarity are addressed differently. In the EPAR of Biograstim®, the mobilisation of peripheral blood progenitor cells is the only area of uncertainty and was due to the lack of complete understanding of the MOA. The case of Biograstim® indicates that there are uncertainties on extrapolation which are tackled in the risk management plan (RMP). In the EPAR of Accofil!®, the totality of quality, non-clinical and clinical data are considered when proving biosimilarity [67].
Safety concerns
In Malta, the Medicines Authority receives feedback from clinicians during annual stakeholder meetings. According to Maltese clinicians, concerns on the safety of biosimilars is one of the barriers to the uptake of biosimilars. For this reason, we investigated this perceived notion by checking for emerging safety concerns by comparing disproportionate adverse event reports pre- and post-biosimilar licensure. Sixteen biosimilar active substances authorized between 2007 and 2019 were evaluated and results are shown in Table 3.
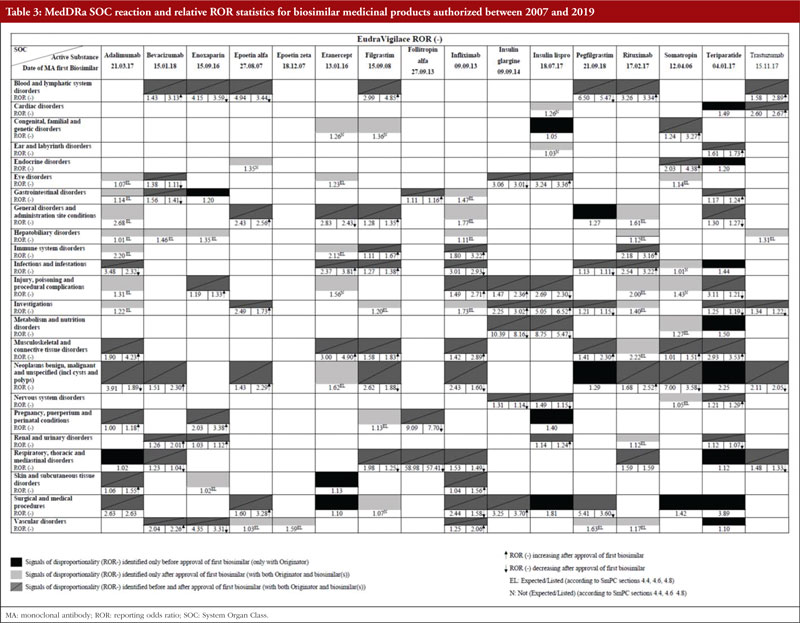
Medical Dictionary for Regulatory Activities (MedDRA) SOC reactions for the 16 biosimilars were retrieved. A total of 144 disproportionality reports (DRs) were observed, 18 of which were only present in the pre-approval phase (only with the originator), 42 of which were only present in the post-approval phase and 84 of which were present in both pre-approval and post-approval phases.
New reports of disproportionality (DR only present in the post-approval phase) were further analysed and out of 42 DRs, 33 were expected/listed while nine were not expected/not listed according to the originator product’s SmPCs. Since 33 DRs were expected/listed, the increase in the number of reports to the point of disproportionality could be explained by constant pharmacovigilance monitoring of biosimilars.
To verify if the safety specification of the biological is changing post-biosimilar licensure, the nine non-expected/non-listed DRs were further analysed. For each SOC DR, the corresponding PTs within the SOC MedDRA hierarchy were retrieved from EVDAS and matched with the EMA list of designated medical events (DMEs). Two potential ‘signals’ that were serious enough to warrant further investigation were identified. These were deafness for insulin lispro and foetal malformation for etanercept. Causality assessment of deafness for Insulin Lispro showed that out of 50 Individual Case Safety Reports (ICSRs), 48 were classified as uncertain, one was classified as unlikely, and one was classified as probable as per the French method. Causality assessment of foetal malformation for etanercept showed that all 226 ICSRs were uncertain (it is pointed out that during the case review, 225 out of 226 ICSRs were found to duplicates of the same case). From this analysis of post-marketing adverse event data, we did not observe any emerging safety concerns related to biosimilars. Since 2005, no new safety issue has arisen when biosimilars have been authorized in line with the EU framework.
Discussion
Since setting up a regulatory framework on biosimilars in 2005, more experience has been gained by the EU regulatory network in approving biosimilar medicinal products, as evidenced by the increased number of marketing authorizations granted over time. Guidelines on the development of specific biosimilars have been described as ‘living documents’ [68] since they are constantly revised and updated as needed.
During the review of EPARs it was observed that, in general, the product-specific guidelines were followed by applicants. There were some cases where some deviations from guidelines occurred, however, approval was still granted since the overall application still showed biosimilarity.
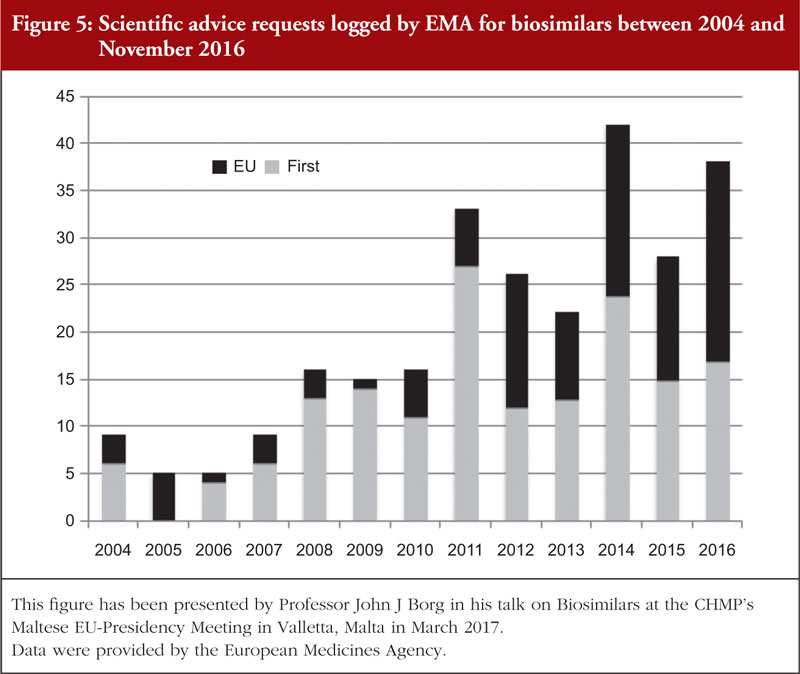
Applicants often sought to engage with the regulators during the product development to obtain scientific advice concerning different quality, non-clinical and clinical aspects of the biosimilar’s development. This is reflected in the number of scientific advice requests which were logged on adapting clinical and chemistry development programmes for biosimilars in one region to meet the EU requirements, see Figure 5. In addition to this, in 2017 the EMA started a pilot project on tailored scientific advice to support companies in carrying out appropriate studies on biosimilars [69].
The review of clinical development programmes of biosimilars currently marketed within the EU showed that the average number of phase I clinical trials was generally higher than the number of phase III clinical trials, except for biosimilars approved in 2016 in which the average number of phase I and phase III clinical trials were the same. It was also observed that, in general, the average number of both phase I and phase III clinical trials decreased in the reviewed period (2007–2019). It was also observed that certain clinical development programmes consisted of only phase I clinical trials without phase III studies. Phase III clinical trials are usually carried out with the intent to support clinical equivalence between the biosimilar and originator following full quality data and phase I clinical trials. In the literature, some authors have argued that an informative and robust CMC package together with meaningful and well-planned phase I clinical trials may reduce the uncertainty on biosimilarity to such an extent that, in certain cases, additional large and expensive phase III clinical trials to confirm biosimilarity may not be needed [70, 71].
Generally, the total number of patients enrolled in phase I clinical trials is lower than the number of patients enrolled in phase III clinical trials, however, there have been cases where the number of patients enrolled in phase I trials was greater than the number of patients enrolled in phase III trials, see Table 2. As already said, some biosimilars were also approved when no phase III clinical trials were conducted, as in the cases of certain Pegfilgrastim biosimilars. However, it was also noted that approval in such cases was dependent on the adequate scientific justification of the absence of phase III studies and, in addition, the scientific advice being sought during the clinical development programme phase.
For phase I clinical trials, mainly healthy volunteers were enrolled. Exceptions to this common trend was observed in the clinical development plan (CDPs) of biosimilars of rituximab, insulin glargine and infliximab. In the clinical development programmes of bevacizumab, etanercept and trastuzumab, it was observed that only healthy male volunteers were enrolled. Under-representation of women in clinical trials is a well-known issue present when studying most medicinal products and is mainly related to the fact that women have childbearing potential and their exclusion from clinical trials is in an attempt to protect fetuses from unknown side effects [72, 73].
Extrapolation of indication was a common approach. Concerns on extrapolation of indication are present since biosimilars can be authorized with the same indications for which the biological originator is approved, without conducting clinical trials with the biosimilar on a specific disease state. Extrapolation for the other indications is permitted if it is properly justified. Regulatory agencies operate strict control when assessing supporting data provided by the applicant for extrapolation of indication [9]. However, extrapolation of an indication remains a challenge from a regulatory perspective, since demonstrating safety and efficacy of a biosimilar is challenging, due to their biological differences. Moreover, each product-specific guideline provides recommendations on studies to be carried out to potentially allow the extrapolation of indication. In certain cases, extrapolation was restricted to some indications as biosimilarity was not shown in all route of administrations. Although extrapolation was based on scientific ground, concerns related to safety of biosimilars are still present in clinical practice.
Results from the analysis of MedDRA SOC reactions showed that 58.3% of SOC reactions occurred in both the pre-approval phase (only with the originator) and in the post-approval phase (following approval of the first biosimilar medicinal product), suggesting a high degree of similarity in the safety profile of biological originators and their biosimilars. Also, from the analysis of PTs, it was observed that 78.6% of the reaction were expected and listed in biosimilar’s SmPCs, therefore are already known and labelled. Among the two non-expected adverse events, it was observed that the PT reactions which matched with the DME list, and hence deserve further analysis, (deafness for insulin lispro and foetal malformation for etanercept) were unlikely to be related to the use of the relevant biosimilar. Deafness has several aetiologies, including diabetes and patients’ ages [74]. In addition, data retrieved from EVDAS did not indicate any causality between the identified PTs and the use of etanercept and insulin glargine biosimilars.
Conclusions
Clinical development programmes of biosimilars and the approval requirements set up by regulatory agencies are changing over time. The EU regulatory framework focuses on CMC and biosimilarity. Therapeutic equivalence is arrived at through PK/PD endpoints, extrapolation, PD markers and PK reliance. This integrated way of proving biosimilarity, reduces the number of clinical trials needed to show biosimilarity and, over time, may lead to shorter clinical development programmes and faster access to medicinal products for patients. Scientific advice is a useful regulatory tool that allow applicants to discuss their development strategies with the regulator early during the development phase, which facilitates the development of safe and efficacious biosimilar medicinal products. Following the regulatory experience of approving biosimilar in the EU, guidelines are being updated to reflect new knowledge on biosimilars gained. Analysis of the spontaneous reports indicates that the above-mentioned framework results in robust quality, safety and efficacy of biosimilar medicinal products, as no increase in safety concerns is observed. These results show that the EU framework of biosimilar is moving in the right direction.
In conclusion, regulation of biosimilars is progressing as more knowledge is being gained and this is reflected in the application of the product-specific guidelines.
For patients
A biosimilar is a medicine which is similar, but not identical, to a reference biological medicine which is already being utilized in clinical practice. Similarity between a biosimilar and a reference biological medicine is demonstrated via physicochemical, clinical safety and clinical efficacy characteristics. In the EU, the regulatory framework for biosimilars is built on guidelines, with EMA issuing the first guideline on similar biological medicinal products in 2005. Since 2005, a total of 12 guidelines have been issued, nine of which were updated in line with new knowledge gained by regulators. Using these guidelines as a direction to be followed, 90 applications for various biosimilars have been submitted by applicants to EMA and 53 biosimilars are marketed in 2019 (biosimilars of adalimumab, bevacizumab, enoxaparin sodium, epoetin, etanercept, filgrastim, follitropin alfa, infliximab, insulin glargine, insulin lispro, pegfilgrastim, rituximab, somatropin, teriparatide, trastuzumab). The safety review carried out does not show emergence of any new adverse events following biosimilars reaching the market. The European framework supports the development and authorization of good quality, safe and efficacious biosimilar medicinal products.
Funding sources
None.
Competing interests: The authors declare no direct or indirect potential conflicts of interest.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Marta Zuccarelli1, PharmD
Benjamin Micallef1, PharmD
Mark Cilia1, PharmD
Professor Anthony Serracino-Inglott1,2, PharmD
Professor John-Joseph Borg1,3, PhD
1Malta Medicines Authority, Sir Temi Żammit Buildings, Malta Life Sciences Park, San Ġwann SĠN 3000, Malta
2Department of Pharmacy, Faculty of Medicine and Surgery, University of Malta, L-Università ta’ Malta Msida, MSD 2080, Malta
3School of Pharmacy, Department of Biology, University of Tor Vergata, Rome, Italy
References
1. The European Parliament and the Council of the European Union. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. OJ L 311, 28.11.2001, p 67.
2. European Medicines Agency. Guideline on similar biological medicinal products. CHMP/437/04 Rev 1. 23 October 2014 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf
3. Challand RH, Gorham H, Constant J. Biosimilars: where we were and where we are. J Biopharm Stat. 2014;24(6):1154-64.
4. Minghetti P, Rocco P, Cilurzo F, Del Vecchio L, Locatelli F. The regulatory framework of biosimilars in the European Union. Drug Discov Today. 2012;17(1-2):63-70.
5. European Medicines Agency. Guideline on similar biological medicinal products. CHMP/437/04. 2005. London, 30 October 2005 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-first-version_en.pdf
6. GaBI Online – Generics and Biosimilars Initiative. EU guidelines for biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2021 Feb 11]. Available from: https://www.gabionline.net/Guidelines/EU-guidelines-for-biosimilars
7. European Medicines Agency. Multidisciplinary: biosimilar [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/multidisciplinary/multidisciplinary-biosimilar
R8. Stevenson JG. Clinical data and regulatory issues of biosimilar products. Am J Manag Care. 2015;21(16 Suppl) s320-30.
9. Tesser JR, Furst DE, Jacobs I. Biosimilars and the extrapolation of indications for inflammatory conditions. Biologics. 2017;11:5-11.
10. European Medicines Agency. Medicines [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines
11. European Medicines Agency. Amgevita. 2017 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/amgevita
12. European Medicines Agency. Imraldi. 2017 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/imraldi
13. European Medicines Agency. Hyrimoz. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/hyrimoz
14. European Medicines Agency. Hefiya. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/hefiya
15. European Medicines Agency. Halimatoz. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/halimatoz
16. European Medicines Agency. Hulio. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/hulio
R17. European Medicines Agency. Idacio. 2019 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/idacio
18. European Medicines Agency. Mvasi. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/myasi
19. European Medicines Agency. Zirabev. 2019 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/myasi
20. European Medicines Agency. Epoetin alfa Hexal. 2007 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/epoetin-alfa-hexal
21. European Medicines Agency. Abseamed. 2007 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/abseamed
22. European Medicines Agency. Binocrit. 2007 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/binocrit
23. European Medicines Agency. Silapo. 2007 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/silapo
24. European Medicines Agency. Retacrit. 2007 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/retacrit
25. European Medicines Agency. Benepali. 2016 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/benepali
26. European Medicines Agency. Erelzi. 2017 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/erelzi
27. European Medicines Agency. Tevagrastim. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/tevagrastim
28. European Medicines Agency. Ratiograstim. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/Ratiograstim
29. European Medicines Agency. Filgrastim Hexal. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/filgrastim-hexal
30. European Medicines Agency. Zarzio. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/zarzio
31. European Medicines Agency. Nivestim. 2010 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/nivestim
32. European Medicines Agency. Grastofil. 2013 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/grastofil
33. European Medicines Agency. Accofil. 2014 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/accofil
34. European Medicines Agency. Ovaleap. 2013 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/ovaleap
35. European Medicines Agency. Bemfola. 2014 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/bemfola
36. European Medicines Agency. Inflectra. 2013 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/inflectra
37. European Medicines Agency. Remsima. 2013 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/remsima
38. European Medicines Agency. Flixabi. 2016 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/flixabi
39. European Medicines Agency. Zessly. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/zessly
40. European Medicines Agency. Abasaglar (previously Abasria). 2014 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/abasaglar-previously-abasria
41. European Medicines Agency. Semglee. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/semglee
42. European Medicines Agency. Pelgraz. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/pelgraz
43. European Medicines Agency. Udenyca. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/udenyca
44. European Medicines Agency. Fulphila. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/ fulphila-0
45. European Medicines Agency. Ziextenzo. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/ziextenzo
46. European Medicines Agency. Pelmeg. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/pelmeg
47. European Medicines Agency. Cegfila (previously Pegfilgrastim Mundipharma). 2019 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/pegfilgrastim-mundipharma
48. European Medicines Agency. Grasustek. 2019 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/grasustek
49. European Medicines Agency. Truxima. 2017 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/truxima
50. European Medicines Agency. Blitzima. 2017 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/blitzima
51. European Medicines Agency. Ritemvia. 2017 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/ritemvia
52. European Medicines Agency. Riximyo. 2017 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/riximyo
53. European Medicines Agency. Rixathon. 2017 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/rixathon
54. European Medicines Agency. Ontruzant. 2017 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/ontruzant
55. European Medicines Agency. Herzuma. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/herzuma
56. European Medicines Agency. Kanjinti. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/kanjinti
57. European Medicines Agency. Trazimera. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/trazimera
58. European Medicines Agency. Ogivri. 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/ogivri
59. European Medicines Agency. Designated Medical Event (DME) list. EMA/326038/2020. 15 June 2020 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/documents/other/designated-medical-event-dme-list_en.xlsx
60. Miremont-Salamé G, Théophile H, Haramburu F, Bégaud B. Causality assessment in pharmacovigilance: the French method and its successive updates. Therapie. 2016;71(2):179-86.
61. European Medicines Agency. Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing recombinant granulocyte-colony stimulating factor. EMEA/CHMP/BMWP/31329/2005, London, UK 2006 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/annex-guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins_en.pdf
62. European Medicines Agency. Guideline on similar biological medicinal products containing recombinant granulocyte-colony stimulating factor (rG-CSF) Draft. EMEA/CHMP/BMWP/31329/2005 Rev 1. 26 July 2018 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-similar-biological-medicinal-products-containing-recombinant-granulocyte-colony_en.pdf
63. European Medicines Agency. Guideline on non-clinical and clinical development of similar biological medicinal products containing low-molecular-weight-heparins. EMEA/CHMP/BMWP/118264/2007, London, UK 2009 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-non-clinical-clinical-development-similar-biological-medicinal-products-containing-low_en-0.pdf
64. European Medicines Agency. Guideline on non-clinical and clinical development of similar biological medicinal products containing low-molecular-weight-heparins. EMEA/CHMP/BMWP/118264/2007 Rev. 1, 10 November 2016 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-non-clinical-clinical-development-similar-biological-medicinal-products-containing-low_en.pdf
65. European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. EMA/CHMP/BMWP/403543/2010. 30 May 2012 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf
66. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005 Rev1. 18 December 2014 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf
67. Cazap E, Jacobs I, McBride A, Popovian R, Sikora K. Global acceptance of biosimilars: importance of regulatory consistency, education and trust. Oncologist. 2018;23(10):1188-98.
68. Wolff-Holz E, Tiitso K, Vleminckx C, Weise M. Evolution of the EU Biosimilar Framework: past and future. BioDrugs. 2019;33(6):621-34.
69. European Medicines Agency. Tailored scientific advice to support step-by-step development of new biosimilars. 16 December 2016 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.ema.europa.eu/en/news/tailored-scientific-advice-support-step-step-development-new-biosimilars
70. Frapaise FX. The end of phase 3 clinical trials in biosimilars development? BioDrugs. 2018;32(4):319-24.
71. Webster CJ, Woollett GR. Comment on “The end of phase 3 clinical trials in biosimilars development?”. BioDrugs. 2018;32(5):519-21.
72. Oertelt-Prigione S. Gender differences and clinical trial design. Clin Invest. 2011;1(2):187-90.
73. Labots G, Jones A, de Visser SJ, Rissmann R, Burggraaf J. Gender differences in clinical registration trials: is there a real problem? Br J Clin Pharmacol. 2018;84(4):700-7.
74. Horikawa C, Kodama S, Tanaka S, Fujihara K, Hirasawa R, Yachi Y, et al. Diabetes and risk of hearing impairment in adults: a meta-analysis. J Clin Endocrinol Metab. 2013;98(1):51-8.
75. U.S. Food and Drug Administration. Biosimilar and interchangeable products. 23 October 2017 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products
76. World Health Organization. Expert Committee on Biological Standardization – Sixtieth Report Annex 2 – Guidelines on evaluation of similar biotherapeutic products (SBPs). Geneva, Switzerland 2009 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.who.int/biologicals/publications/trs/areas/biological_therapeutics/TRS_977_Annex_2.pdf?ua=
77. Health Canada. Biosimilar biologic drugs in Canada: Fact sheet. 23 August 2019 [homepage on the Internet]. [cited 2021 Feb 11]. Available from: https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/dhp-mps/alt_formats/pdf/brgtherap/applic-demande/guides/Fact-Sheet-EN-2019-08-23.pdf
78. Chow SC. Biosimilars: design and analysis of follow-on biologics. Chapman and Hall; 2014. p. 55.
79. Choy E, Jacobs IA. Biosimilar safety considerations in clinical practice. Semin Oncol. 2014;41 Suppl 1:S3-14.
|
Author for correspondence: Professor John-Joseph Borg, Post-Licensing Directorate, Medicines Authority, Sir Temi .ammit Buildings, Malta Life Sciences Park, San .wann, S.N 3000, Malta |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2021 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


