1st ASEAN overview workshop on GMP for biologicals/biosimilars 2018 – Report
Published on 2014/03/20
Generics and Biosimilars Initiative Journal (GaBI Journal). 2019;8(3):119-27.
|
Introduction: The Association of Southeast Asian Nations (ASEAN) Overview Workshop on GMP for Biologicals and Biosimilars was co-organized with the Generics and Biosimilars Initiative (GaBI). This meeting was intended to improve the understanding of the good manufacture practice (GMP) inspection framework for biological (and biosimilar) drugs among ASEAN countries. |
Submitted: 18 February 2019; Revised: 24 July 2019; Accepted: 25 July 2019; Published online first: 6 August 2019
Introduction
In collaboration with the Association of Southeast Asian Nations (ASEAN), the Generics and Biosimilars Initiative (GaBI) organized a first workshop of its kind in this region on good manufacturing practices (GMP) for biological drugs (including biosimilars).
The workshop held on 5 August 2018 focused on GMP standards and the knowledge required for effective GMP inspection of biological products. In addition, there were discussion sessions to identify common concerns in these areas.
It was attended by the ASEAN Joint Sectoral Committee (JSC) on GMP inspection, other ASEAN GMP inspectors, reviewers from the ACCSQ-PPWG (ASEAN Consultative Committee for Standards and Quality-Pharmaceutical Product Working Group) Member States, academics, regulators and other experts, including from the World Health Organization (WHO). In total, 46 people attended the workshop, which included a series of presentations, each followed by a Q&A session, and parallel group discussions.
Results/Processes
Expert speaker presentations
There were a number of expert speaker presentations followed by Q&A and an in-depth panel discussion. The presentations are downloadable from the GaBI website [1].
The workshop began with a welcoming speech from Mr Do Van Dong, Deputy Director General of the Drug Administration of Vietnam.
Mr Dong discussed the importance of GMP for the entire lifecycle of biological and biosimilar products. He noted that WHO has published several different versions of its guidelines on GMP practices and principles and recently updated its specific requirements. The latest version of WHO GMP for biologicals and biosimilars (WHO Technical Report Series (TRS) No. 999, Annex 2 [2]) replaces the previous version from 1992. However, Mr Dong highlighted the disproportionate input from the Western world on GMP practices. As a result, the ASEAN Member States (AMS) lack opportunities to update their GMP practices, especially for biological products. Mr Dong stated that the present workshop was an encouraging start for the development of an ongoing forum for the exchange of knowledge and experience among GMP inspectors, as well as for the promotion of consistency and harmonization of the relations among the regulatory authorities within the ASEAN community with regards to GMP for biologicals.
GMP for biologicals and biosimilars – workshop objectives
Chair of the ASEAN Joint Sectoral Committee on GMP Inspection, Adjunct Associate Professor Chong Hock Sia then provided a second welcome address and outlined of the workshop objectives.
Professor Sia introduced ASEAN and noted that its Member States have a combined population of 650 million people and a combined economy of US$2.5 trillion. He also discussed aspects of the ASEAN Mutual Recognition Agreement (MRA) on GMP inspection [3] but noted that this does not yet include biologicals, active pharmaceutical ingredients (APIs) or investigational medicinal products. As such, he highlighted the importance of the workshop and its objectives:
1) To understand current GMP (cGMP) inspection framework for biologicals/biosimilars based on Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (PIC/S) or other equivalent cGMP standards
2) To promote active discussion amongst inspectors, reviewers and scientists from AMS concerning best practices to use when inspecting manufacturers of biologicals/biosimilars
3) To identify areas of consensus, uncertainty or disagreement concerning inspection framework on cGMP for biologicals/biosimilars.
Overall, the workshop aimed to increase the ability of participants to conduct inspection of biological/biosimilar manufacturing facilities under the cGMP framework, a critical component of the registration and licensing of pharmaceutical and biological products.
Newly revised WHO GMP for biological products
Dr Dianliang Lei, a WHO scientist in Technologies, Standards and Norms for Essential Medicines and Health Products, discussed the 2015 updated WHO GMP guidance for biologicals.
Dr Lei explained that WHO’s GMP guidelines for biologicals were first established in 1992. These have now been revised and updated in accordance with the revision principles [2], see Table 1. A number of challenges were encountered during the revision process but key changes to the guidelines have now been made. These include changes to the scope, terminology, pharmaceutical quality and risk control, starting materials, campaign production, containment, labelling and documentation. Containment and biosafety were also introduced into the GMP guideline. Measures such as segregation of live and non-live material, clothing, product and material transfer procedures, HVAC design, acquiring specific knowledge on the type of microorganisms being handled and its associated risks, environmental monitoring tailored based on the risks and characteristics of the biological products, and the use of a campaign-based production considering upstream and downstream processes are addressed in the guidelines. Terms such as reference sample and retention sample were used in the revised GMP guidelines. Reference sample is a sample of a batch of starting material, packaging material, intermediate or finished product which is stored for the purpose of being analysed should the need arise during the shelf-life of the batch concerned. Retention sample is a sample of a fully packaged unit from a batch of finished product. It is stored for identification purposes. In certain situations, both samples are exchangeable.

Characterization and testing of animal cell substrates used for production of biologicals
Dr Elwyn Griffiths, former Chairman (2010–2016) and Rapporteur (2016–2017) of the WHO Expert Committee on Biological Standardization, and workshop Chair, discussed the use of animal cells in the production of biological drugs and vaccines. Cell banks are critical to the production of modern biological medicines, Some of which were previously extracted from animal fluids or tissues, the range of cells used include microbial, animal and plant cells.
Dr Griffiths outlined some of the critical manufacturing issues in producing biological drugs from cells, including the production system; the genetic stability of the cell substrate and microbial seeds; viral safety issues and impurities caused by the host cell such as residual deoxyribonucleic acid (rDNA) from mammalian cells, see Table 2. He stressed that small process changes can have a major impact on the clinical performance and safety of a biological product, making production consistency critical.

He noted that continuous mammalian cell lines are the substrate of choice for many rDNA products as they can be transfected and engineered to grow rapidly and produce glycosylated products. He outlined the three major concerns associated with using these cells to produce biological drugs:
1) Genetic stability of cell lines
2) Residual host cell DNA, which might cause cancer
3) Viruses in animal cells, including re-troviruses
Dr Griffiths introduced the concept of a Master Cell Bank (MCB, cells derived from a cell seed and frozen) and Working Cell Bank (WCB, derived from the MCB and used to provide cells for manufacturing) which provide a standardized source of production cells and are now used for all cell lines. To prevent viral contamination, WHO encourages production based on cryopreserved cell banks exhaustively screened for virus contamination; control of raw material used in production; closed systems of cell culture; testing each cell culture for contamination, and validation of viral removal/inactivation by downstream processing.
Dr Griffiths noted that cell banks require highly specialized expertise and infrastructure, and this is often contracted out to specialized testing organizations. He described good cell culture practice, including ensuring the donor is free of transmissible diseases, and confirming the absence of viral and microbial contamination. The MCB, WCB and all cell culture processes are key to consistently producing safe and effective biologicals.
Fermentation: fundamentals, control of source materials and cell culture conditions
Focusing on fermentation, Dr Dinesh Khokal of Amgen Singapore, first described the upstream and downstream processes for biological manufacturing, see Figure 1.
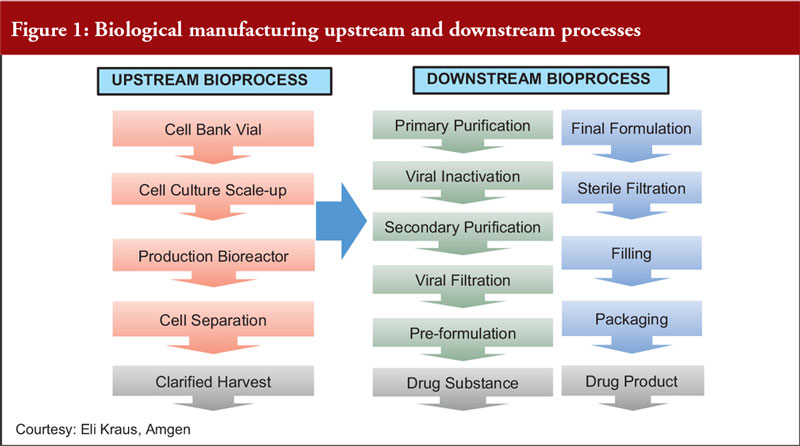
Dr Khokal explained the importance of process control during fermentation. He noted that cell culture contamination is the most common problem associated with fermentation. This may arise due to impurities in the source materials or biological contaminants such as unwanted bacteria, viruses or moulds. To reduce the risk of contamination, he made recommendations including:
- The use of pumps to move fluid; filters to guard against impurities; and/or equipment to provide ultrapure oxygen, carbon dioxide, water for injection and ingredients for the fermentation medium
- Sterilization of filter inlet and exhaust gas
- Filtration of the culture medium
- Only allowing contact of sterile materials with sterile instruments
- Minimizing personal intervention and provision of appropriate training to all personnel
- Employing a personnel monitoring programme, including monitoring of operator’s gloves on a daily basis
Dr Khokal also emphasized the importance of control of the source materials in order to reduce the risk of adventitious agent contamination and other serious loss of quality and safety. It is important to ensure the origin and quality of source materials according to GMP principles and use a sampling, testing and monitoring programme. He also recommended aseptic manufacturing processes and controlled transportation of critical materials to manufacturing sites.
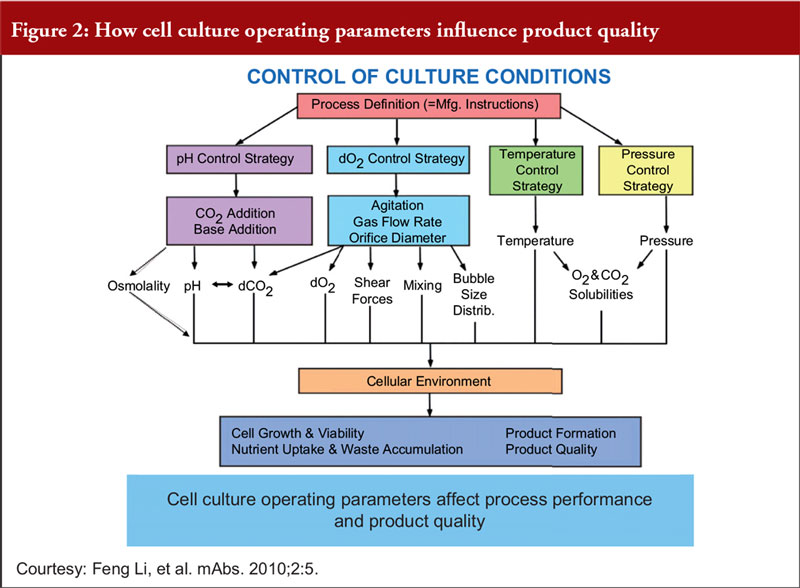
Finally, Dr Khokal explained that even slight changes to the culture conditions can impact culture performance, productivity and product quality, see Figure 2. He added that fermentation processes (including sterile practices, control of source materials and culture conditions) should be risk-based, science-based and in accordance with WHO GMP guidelines for biological products.
Purification of vaccines and biologicals
Commercial Research Manager of Indian biotechnology firm Protaccine Biotec, Dr Anil Kumar Chawla, discussed how to purify biological drugs, including vaccines. He began by explaining that the first step in the manufacturing process — the harvesting/purification of a protein from the reactor — introduces a significant risk of product degradation, bioburden concerns and/or process errors, see Table 3.

He outlined the two major techniques for purification, which aim to generate a highly pure active drug substance free of all possible impurities:
1) Cell disruption
2) Separation of soluble products
In conclusion, Dr Chawla highlighted the importance of quality risk management (QRM) and made some critical observations on WHO inspections. He noted that manufacturers should control the bioburden during purification, carefully store purification equipment, use disposable accessories wherever possible, monitor clean room parameters, replace aseptic processes with sterile filtration and improve the purification process based on historical data.
Harvest process in commercial biologicals manufacturing
Dr Yusdy Pan, Principal Scientist for Process Development for Amgen Singapore, outlined the harvest process for biological drugs, including new harvest technologies for high density cell culture.
Dr Pan outlined the differences between mammalian and microbial cell expression systems, see Figure 3.
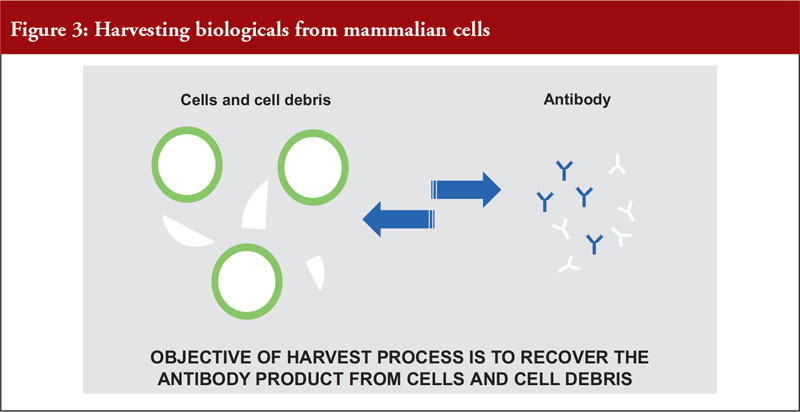
He also noted that mammalian cell expression systems offer a simple antibody recovery system, which is what is harvested. The conventional harvest process for mammalian cell culture involves a combination of centrifugation and filtration and is suitable for a cell density of a maximum of 10 million cells per mL, see Figure 4.
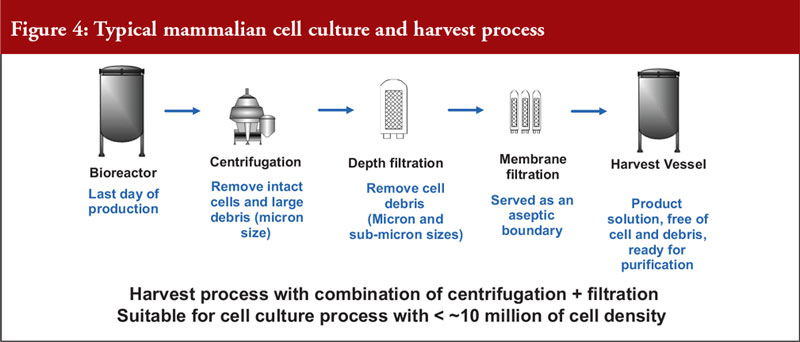
To meet the industrial demand for new high-density cell culture techniques, which can involve culturing over 10 million cells and a solid content of over 15%, alternative technologies are needed. The conventional platform process is unsuitable for this due to limited centrifuge bowl capacity and depth filter surface area. Alternative technologies include:
- Accelerated sedimentation (flocculation)
- Tangential flow-microfiltration
- Alluvial filtration
- Cadence acoustic separation
How to build an effective pharmaceutical quality system
Mr Vimal Sachdeva, Senior Inspector in the WHO Prequalification Team, explained the WHO Prequalification of Medicines Programme.
He introduced the quality management system (QMS) as important to facilitate innovation and continual improvement and to strengthen the link between pharmaceutical development and manufacturing activities. He said a streamlined structure enables compliance and operational efficiency and the flexibility to incorporate different modalities.
With regards to this, WHO states that all biological products must be manufactured in accordance with pharmaceutical quality system (PQS) requirements, as defined in WHO GMP. QRM principles must also be used to develop control strategy across all stages of the manufacturing process, which should involve ongoing trend analysis and periodic review, starting material control and change control. It is also important to design monitoring systems and a control strategy to manage any identified risks. The International Council for Harmonization (ICH) Q10 pharmaceutical quality system aims to promote a move away from discrete GMP compliance procedures to a comprehensive quality systems approach, across the whole lifecycle of the product.
Mr Sachdeva gave examples of effective QRM and examples where the implementation of QMS had failed. He then concluded that biological quality objectives should be clearly linked to business objectives and strategy and the QMS should be clear and establish the link between quality policies and their implementation on the ground. To adapt to changes in regulation and incorporate site-specific nuances, the QMS should be flexible. Manufacturers need to ensure that all elements of the QMS are well connected and use contract manufacturing organizations where necessary; and use knowledge to improve processes. Finally, he noted that there should be an effective, proactive QRM process including internal audits and product quality reviews, see Table 4 on the general principles of a good QRM.

Validation of viral removal/inactivation and bioanalytical methods
Dr Dinesh Khokal spoke about the removal of viral contamination from biological products. This is an important issue as viral contamination is a risk to all biological drugs. Contamination can arise from the original source of the cell lines, or from the adventitious introduction of a virus during production. A number of biological drugs have been contaminated in the past and only identified years after manufacture, causing great potential risk to patients. For the manufacturer, this can lead to facility shutdowns and significant business impact, see Figure 5 on the potential source of virus contamination.

There are many viral risk mitigation strategies, but none of these alone can provide a high enough level of assurance so they must be used in combination. Furthermore, because no single test can test for all known viruses, validation of the process for viral removal or inactivation is essential in establishing product safety. Methods to achieve this include viral inactivation and viral removal. These methods must be validated.
Dr Khokal discussed the importance of choosing which viruses should be used in viral clearance studies, including relevant viruses and model viruses. He also described the various assay types and noted that any assay must produce accurate measures of the viral load (usually expressed with 95% confidence limit, around 5% log of the mean).
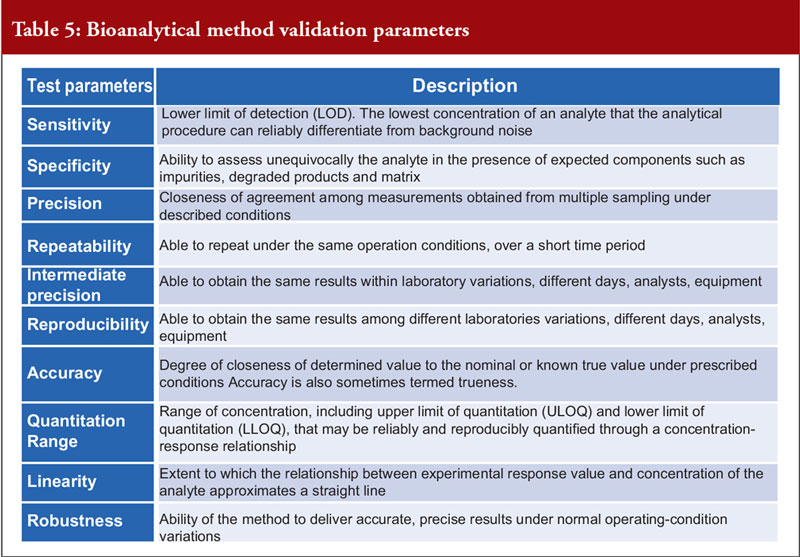
Dr Khokal concluded by explaining that validation of the bioanalytical method used is important to assess that the method is fit-for-purpose, to ensure that the data are reliable to support the safety and effectiveness of the biological and critical for the quantitation of analytes (including biological products) and biomarkers in biological samples. There are several important method validation parameters, see Table 5. He noted that the US Food and Drug Administration (FDA) offers guidance on bioanalytical method validation, which outlines in depth the parameters required for chromatographic and ligand binding assays.
Batching and storage of bulk biological products
Dr Anil Kumar Chawla discussed the process of batching bulk biological products. Biological products can be manufactured in sublots and pooled as one batch, which means the bulk batch can be packed differently in terms of volume/concentration. He explained that the batch number should be recorded according to quantity, manufacturing date, expiry date, strength and excipient(s). Here, Dr Chawla noted that traceability is the key concern.
He also said the batch numbering standard operating procedure (SOP) should identify risks such as from inadequate numbering and mix-ups, clearly differentiable batch numbers should be used and risk scores should be calculated for each batch numbering system.
Dr Chawla outlined a number of critical criteria for bulk batching during WHO inspections, including:
- Traceability of the finished product from bulk batches
- Consistency of batch numbers
- Stability studies of bulk biological products
- Media simulation studies for aseptic processes
- Material stores for bulk products, sublots or intermediates
- Holding time studies for bulk materials
- Temperature monitoring in storage areas
- Compatibility of labels to storage temperature
- Container-closure integrity during storage and transit
- Studies on container-closure systems for bulk products
Moving on to the storage of bulk products, Dr Chawla explained that the primary container should protect the bulk product from the external environment, keep it stable and be of appropriate material. He outlined the necessary aspects of storage conditions which included temperature, material, location and sanitation. He also outlined the requirements for distribution and the monitoring of transportation. And he concluded by noting the importance of monitoring extreme environmental conditions, specifically that special temperature monitoring systems and alert mechanisms should be used.
Data integrity, from an inspector’s point of view
Mr Vimal Sachdeva introduced the importance of data integrity in biological production, with reference to WHO guidance on this issue.
He noted that data integrity means ensuring data are recorded as intended and the same in content and meaning as when it was originally recorded at all times, as well as preventing unintended or unauthorised changes to data. Data integrity is the degree to which a collection of data is Attributable Legible Contemporaneous Original and Accurate (ALCOA).
Accurate scientific data are critical as the basis of risk/benefit decisions regarding the selection and use of drugs. This is vital to avoid harm to patients and retain trust in the effectiveness of products and those that supply them.
He quoted WHO’s expectations on data management, which state that ‘good data and record management are critical elements of the pharmaceutical quality system and a systematic approach should be implemented to provide a high level of assurance that across the product life-cycle all GMP records and data are accurate, consistent, trustworthy and reliable’. Many data regulations exist across the globe.
Mr Sachdeva gave multiple examples of real-life data integrity breaches, including one case where plates recorded and reported as negative by quality control (QC) personnel were actually positive for contamination. He noted that there can be various causes of data integrity breaches, that include human error, system level issues and technology issues.
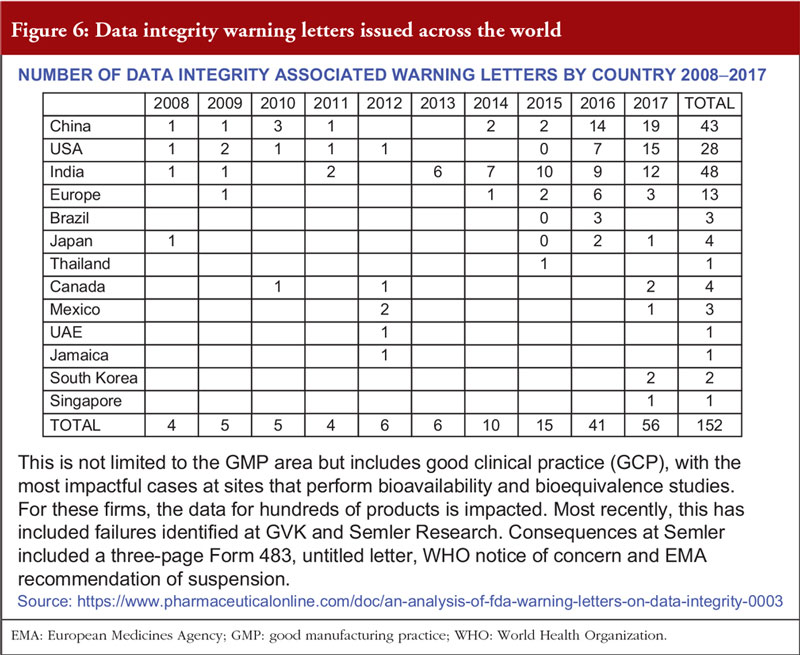
Overall, the presentation suggested that data integrity issues are increasing, see Figure 6. Often, inspections reveal deficiencies due to non-compliance with GMP. Mr Sachdeva concluded that data integrity issues are corrosive to science and trust, which – once lost – cannot be restored.
WHO recommendations on cell substrates used for production
Speaking for a second time, Dr Dianliang Lei presented the WHO’s requirements for the cells used to manufacture biologicals, which can affect the characteristics and safety of biological products.
The potential risks of using cell substrates include viruses and other infectious agents, cellular DNA and ribonucleic acid (RNA), and growth promoting (oncogenic) proteins. A two-tiered cell bank system can be used to avoid genetic drift and limit the number of paths necessary. Dr Lei outlined the different types of animal cell substrates and their advantages and disadvantages, they are primary cell culture, diploid cell lines and continuous cell lines.
When developing cell cultures, fundamental features include authenticity (identity/provenance), the absence of contamination (with another cell line/microbes) and stability. GMP provides guidance on establishment of the cell bank, traceability to the originator cell line, storage, handling, cross-contamination and adventitious agent contamination. WHO requirements on cell banks include GMP for Biologicals (TRS 999), GMP for APIs (TRS 957) which is the specific guidance for APIs manufactured by cell culture or fermentations, recommendations for cell substrates (TRS 978) and other product specific recommendations. WHO’s recommendations for cell substrates is limited to animal cells but does consider novel substrates, including cells of avian, canine and insect origin. Issues related to stem cell lines for biological production are included, but not stem cells for therapy by transplantation.
Dr Lei ended by noting that the appropriateness of continuous cell lines for the biological product must be considered, and the MCB and WCB must be fully characterized. Cell bank characterization should include preliminary evaluation (such as for oncogenicity). WHO provides a summary of testing for the evaluation and characterization of the MCB and WCB in its revised draft recommendations, based on input from manufacturers (TRS 978 Annex 3).
Importance of GMP in controlling cell substrates and production processes for biologicals – two case studies
Chair Dr Elwyn Griffiths closed the session by providing two case studies to illustrate the importance of following GMP in biologicals production. He noted that consistency of production is critical and depends on factors including the cell substrate, cell production system and separation/purification method.
Viral contamination is a key issue in the safety of biological products. Dr Griffiths noted that guidelines, e.g. WHO recommendations for the evaluation of animal cell cultures as substrates for the manufacture of biological products and for the characterization of cell banks, consider the possible viral contamination of live viral vaccines and rDNA products produced in any mammalian cell as a major issue. Guidance is regularly updated to account for new scientific information and technologies.
He also noted that the MCB and WCB should be exhaustively screened for viral contamination and the raw material used in production including growth media and enzymes should be controlled. Closed systems of growth and testing of each cell culture lot should be employed. Finally, validation of viral removal/inactivation by downstream processing is key. In cases where viral contamination does occur, how it is handled is critical.
To illustrate this last point, he gave two real-world examples of viral contamination, with opposing outcomes. The Genentech’s contamination by minute virus of mice (MVM) in 1993 and 1994 and Genzyme’s Vesivirus contamination in the early 2000s. One situation led to global drug supply being compromised and to the near collapse of the company, demonstrating the seriousness of viral contamination. The other was handled successfully.
In conclusion, Dr Griffiths reiterated that manufacturers must deal promptly with any suspected contamination. National regulatory authorities have a critical role in overseeing these developments, but ultimately the continued vigilance of manufacturers is essential.
Summary of the discussions that followed the expert presentations
After the presentations, there was the opportunity for discussion about the topics covered. The key discussion points are summarized below.
WHO GMP for biologicals: input into the process and specificity of guidelines
Following the discussion of revised WHO GMP for biological products, it was emphasized that the guidance is developed not only by WHO but also based on global consultation and significant input from industry and regulators. The guidelines also refer to other compatible guidelines, such as those from FDA, the European Medicines Agency (EMA) and also PIC/S.
There was a further question about extending the guidelines, as biological products are wide-ranging including, for example, recombinant products, vaccines, monoclonal antibodies, yet current GMP standards are not specific to each individual category of product. An audience member suggested this can make it difficult for a manufacturer to navigate the guidelines as they need to look for supplementary guidelines/annexes specific to their product.
The idea of an ‘aide memoir’ for different product categories was discussed, to help remind inspectors which areas to cover during an inspection. However, it was also acknowledged that there are many different ways of carrying out an inspection. A WHO ‘Questions and Answers’ document was also mentioned, which helps to clarify certain aspects of the GMP guidelines.
Host-cell impurities
Following Dr Elwyn Griffiths presentation on the characterization and testing of animal cell substrates, a question was asked regarding host cell-related impurity – as well as DNA which has a limit of 10 ng/dose, the host cell protein can also be problematic because it can cause allergic reactions, so what is the acceptable level of host cell protein.
Dr Griffiths responded that protein contamination has been considered less of an issue than DNA. He also mentioned that it is important for inspectors to properly check how the cell bank has been prepared and the type of facility the manufacturer has used, as any problems with the cell bank will get translated into the product. He reiterated the importance that every vial in the cell bank is consistent.
Cell viability
The level of cell viability required for production was discussed. Dr Yusdy Pan discussed a 30% minimum level for viability. Any less than that can be dangerous, because when many cells die and break down it is more challenging to remove the host cell protein.
Dr Dinesh Khokal made the point that the viability level also depends on the cell line; for example, human cell lines can be more difficult to use than established animal cell lines. It can also depend on the method used to check viability. A higher level of viability is better, but this can be challenging when growing cells at high density using high throughput media.
The speakers also discussed the importance of knowing the product, highlighting that even small changes to increase expression such as a change of media can lead to a change in the end product, for example, by changing protein post-translational modifications. Consistent manufacturing processes are very important to ensure the consistency and quality of the product.
Sustainability of the fermentation process
How disposable technology is overtaking stainless steel in the biological drug industry was discussed. An increasing number of companies that use mammalian cells are moving towards disposable technology. A question was asked about the sustainability implications of single-use technology such as disposable columns and bioreactors.
Dr Khokal responded that manufacturers must comply with the local and environmental ministry’s requirements, which are usually incineration. He also said that the use of disposable technologies is environmentally beneficial in other ways, such as by reducing water and chemical detergent use. He said there is a balance to be struck between disposing plastics versus the volume of water used, power requirements, space and mobility. A further important issue is carryover contamination from a previous product, which is ameliorated by the use of disposable bioreactors.
Column purification
On the issue of purification, it was asked how to ensure that there is no residual product contamination when performing chromatography purification (using the same column with a different product).
Dr Anil Kuwar Chawla responded that it is important to demonstrate that the previous product has been eliminated from both the column and the environment. If purifying a live product, e.g. a virus, in a column you can use the culture method to demonstrate this, but if purifying an inactive product, e.g. a protein, a more sensitive method is necessary. He said it does not align with GMP to use the same regime for multiple different products.
Data integrity
Finally, following Mr Vimal Sachdeva’s session on data integrity, the differences between biological drugs and conventional APIs were discussed. On this issue, Mr Sachdeva said that the vaccine programme is the oldest programme in WHO prequalification of medicines programme, although inspections of vaccine manufacturers are newer.
Data integrity inspections of vaccine manufacturers began three or four years ago and in 2017, two or three sites (out of 10) have had data integrity issues. However, in past experience there have been more data integrity issues with small molecules.
Dr Dinesh Khokal mentioned ‘data integrity challenge exercises’, which are used as a reviewer training exercise. It was also emphasized that senior management has the ultimate responsibility for data integrity, but that data integrity is an issue throughout the manufacturing process – not only when performing analysis. Overall, the manufacturer must have a robust internal audit programme in place which accounts for data integrity.
Action points highlighted in the discussions
The workshop included four parallel group discussions, which had several important outcomes. These are summarized below.
Group 1: WHO or PIC/S and other equivalent international GMP standards
Group 1 discussed the information required for effective GMP inspection.
Action points:
- Although WHO GMP guidelines are not a mandatory requirement (unlike PIC/S GMP), they are useful and should be referred to.
- Improve experience in upstream biologicals manufacturing so that it is equivalent to that in downstream production and quality control testing.
- Share Information related to new technology.
- Introduce bi-regional training, especially on meeting data integrity req-uirements.
- English translation during inspection of non-English speaking manufacturers is needed; help with interpretation of the guidelines; and group of relevant WHO TRS to make them more user-friendly.
Group 2: Difficulties in GMP inspection
Group 2 discussed the difficulties associated with GMP inspection, especially regarding the effect of multiple inspections and a lack of mutual reliance, see Figure 7.

Action points:
- Align the perspectives of industry and regulators on GMP inspection.
- Reduce the differences in the competency of inspectors, differences in regulatory frameworks, lack of communication between regulators and regulation not keeping pace with new technologies.
- Improve the ASEAN MRA, so that it includes biologicals.
- Use external experts with no conflict of interest and use consultants who are not associated with any single association.
- Reduce onsite audits by performing desktop assessments beforehand.
- Enhance communication and education.
Group 3: Viral safety
Group 3 discussed the viral contamination of biological and biosimilar drugs; the following action points were raised.
Action points:
- Ensure inspectors are experts in virology.
- Improve knowledge of the origin of the material.
- Ensure expertise in viral safety to assess the data integrity of test results.
- Ensure inspectors can properly assess the risk of viral contamination and if they cannot at present, they should be trained.
- Ensure that challenges such as emerging viruses from new cell lines, assessing the ability of a contract laboratory to carry out testing and identifying the source of the contamination, can be tackled.
- Increase regional collaboration, i.e. ASEAN-wide initiatives, for the optimum use of resources as well as input from more experienced countries.
- Include reviewers as part of the inspection team.
- Increase training opportunities for inspectors.
Group 4: Processing into finished dosage forms
Group 4 focused on filling and processing biological drugs into finished dosage forms.
Action points:
- Address filtration issues with new technology or approval of products without high level of filtration.
- Meet the need for containers that are compatible with various types of product, which each have different storage requirements, and uniform quality primary packaging materials.
- Get more information from manufacturers and packaging material suppliers.
- Get information and more validation and stability studies.
- Get more information on the impact of external scratches on vials on the product quality. It is currently unclear whether to reject or accept scratched vials.
- Increase the validation studies of lyophilization process and access to production development data. Get more guidance on the lyophilization process from manufacturers.
- Get more compatibility studies for products with heat-sterilized glass vials.
- Increase the safety data on overages (product which contains a concentration beyond a certain limit) being injected into a subject. More preclinical and clinical studies are needed.
Conclusion
The workshop was successful in bringing members of ASEAN together with experts from other nations, to discuss GMP for Biologicals and Biosimilars and improve understanding of the GMP inspection framework for biological (and biosimilar) drugs among ASEAN Member States. It highlighted many important issues surrounding GMP for biological and biosimilar drugs manufactured in ASEAN countries, including the challenges of adhering to general GMP standards, given the huge diversity of biological products; protecting the final product from host cell impurities including DNA and protein; as well as adventitious viral contamination and the importance of having robust quality control programmes in place at all levels of production. Overall, the meeting helped to clarify WHO’s requirements for GMP production of biological drugs and how manufacturers can ensure these standards are met to ensure their product is safe, effective and of high quality. It was an important step forward as the first meeting of its kind for ASEAN Member States.
Speaker Faculty and Moderators
Speakers
Anil Kumar Chawla, PhD, India/Switzerland
Elwyn Griffiths, DSc, PhD, UK
Dinesh Khokal, PhD, Singapore
Dianliang Lei, PhD, Switzerland
Yusdy Pan, PhD, Singapore
Vimal Sachdeva, MSc, Switzerland
Adjunct Associate Professor Chong Hock Sia, BPharm, MSc, Singapore
Moderators
Kakkanang Porkaew, Thailand
Wiwin Wisma Prihatin, Indonesia
Seok Hui Teo, Singapore
Prapassorn Thanaphollert, BS, Thailand
Editor’s comment
Speakers and moderators had provided the discussion/conclusion of the group discussion, read the report and revised the content of the summary discussion.
Acknowledgement
The Generics and Biosimilars Initiative (GaBI) wishes to thank Ms Sylvia Laksmi Sardy and Ms B Lusia Herwahyu S from the ASEAN Secretariat for their support to the organization of this workshop; the moderators in clarifying the information of the parallel discussion when finalizing the meeting report; as well as Dr Elwyn Griffiths and Dr Dianliang Lei, Chair and Co-chair of the 2018 workshop, as well as Adjunct Associate Professor Chong Hock Sia for their strong support through the offering of advice and information during the preparation of the workshop.
The authors would like to acknowledge the help of all the workshop speaker faculty and participants, each of whom contributed to the success of the workshop and the content of this report, as well as the support of the moderators and co-moderators in facilitating meaningful discussion during the parallel case study working sessions, presenting the discussion findings at the meeting, and contributing in the finalization of this meeting report.
Lastly, the authors wish to thank Ms Alice Rolandini Jensen, GaBI Journal Editor, in preparing and finalizing this meeting report manuscript and providing English editing support on the group summaries.
Competing interests: The workshop was sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Elwyn Griffiths, DSc, PhD
Adjunct Associate Professor Chong Hock Sia, BPharm, MSc
References
1. Generics and Biosimilars Initiative. 1st ASEAN Overview Workshop on GMP for Biologicals/Biosimilars 2018. 5 August 2018; Da Nang, Vietnam. Available from: www.gabi-journal.net/about-gabi/educational-workshops/1st-asean-overview-work-shop-on-gmp-for-biologicals-biosimilars-2018
2. World Health Organization. WHO good manufacturing practices for biological Products, WHO Technical Report Series, No. 999, Annex 2 (2016) [homepage on the Internet]. [cited 2019 Jul 24]. Available from: https://www.who.int/biologicals/areas/vaccines/Annex_2_WHO_Good_manufacturing_practices_for_biological_products.pdf
3. ASEAN sectoral mutual recognition arrangement for good manufacturing practice (GMP) inspection of manufacturers of medicinal products [homepage on the Internet]. [cited 2019 Jul 24]. Available from: https://www.asean.org/uploads/archive/22481.pdf
|
Author for correspondence: Adjunct Associate Professor Chong Hock Sia, Senior Consultant (Audit and Licensing) and Director (Quality Assurance), Health Products Regulation Group, Health Sciences Authority Singapore, 11 Biopolis Way, Helios, #11-01, Singapore 138667 |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2019 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


