A bioequivalence study of paliperidone palmitate once-monthly (156 mg/mL) extended-release injectable in patients with schizophrenia
Published on 16 December 2025
Generics and Biosimilars Initiative Journal (GaBI Journal). 2026;15(1).
Study objective: To evaluate the steady-state pharmacokinetic bioequivalence of Mylan’s paliperidone palmitate extended-release injectable suspension 156 mg/mL (test formulation) with Janssen’s Invega Sustenna (R) (paliperidone palmitate) extended-release injectable suspension 156 mg/mL (reference formulation), in patients with schizophrenia, and to evaluate the safety and tolerability of the test formulation. |
Introduction
Schizophrenia is a significant contributor to disability globally and imposes a substantial burden on healthcare systems [1, 2]. The progression of schizophrenia is not only determined by intrinsic factors but can also be influenced by several external factors such as non-adherence to treatment, poor lifestyle, stress, lack of support, and substance use [2, 3].
Non-adherence to treatment is common among patients with schizophrenia, with estimates indicating that 40% to 60% of patients are either poorly adherent or non-adherent to oral antipsychotic (OA) therapy [4]. Non-adherence significantly contributes to relapse, which accelerates disease progression. Relapse prevention may therefore be considered a central goal in schizophrenia management. To add to this, clinicians often lack reliable tools to accurately assess a patient’s adherence to treatment [2, 4, 5].
Long-acting injectable (LAI) antipsychotics are associated with greater adherence and lower rates of relapse, hospitalization, mortality, and suicidal behavior, in comparison to OAS [4, 6]. LAIs could potentially provide better clinical outcomes in patients with existing or anticipated risk of non-adherence to OAS [7]. Additionally, LAIs are effective in treating acute schizophrenia, can help manage psychopathology, and improve adherence early on, from first-episode psychosis [8, 9].
Despite these advantages, LAIs are not widely prescribed, which could be attributed to factors like limited access, minimal knowledge and/or misconceptions among physicians, patients, and caregivers. While LAIs are often reserved for patients with severe symptoms or adherence issues [4], clinical practice guidelines also recommend use of LAIs earlier in the treatment course [10, 11].
Paliperidone palmitate is an atypical antipsychotic indicated in the treatment of schizophrenia and schizoaffective disorder as monotherapy, and as an adjunct to mood stabilizers or antidepressants [3, 11]. It is recommended by various clinical practice guidelines for the treatment of schizophrenia [3, 11]. Development of generics of this complex LAI formulation may provide greater choice to healthcare systems and improve access to patients, while maintaining efficacy and safety [5].
To date, therapeutically equivalent generics are available only for a few LAIs due to technical challenges in their development and strict regulatory requirements [5]. LAIs such as paliperidone palmitate extended-release (PP1M) are classified by the US Food and Drug Administration (FDA) as complex formulations, and it mandates steady-state bioequivalence (BE) studies in patients with schizophrenia or schizoaffective disorder [12]. Parallel group designs are recommended over crossover designs because of PP1M’s long apparent half-life, but these studies demand large sample sizes, multicentre recruitment, and extended treatment durations to achieve steady state. Operational hurdles, such as frequent blood sampling, patient adherence, and high variability further complicate BE demonstration, explaining the scarcity of published studies. This study aims to demonstrate the BE of generic PP1M injectable suspension, 156 mg/mL, in comparison to the reference standard.
Study objective
The primary objective of this study was to evaluate the steady-state pharmacokinetic BE of Mylan’s PP1M injectable suspension 156 mg/mL (test formulation) with Janssen’s Invega Sustenna® (paliperidone palmitate extended-release [PP1M] injectable suspension 156 mg/mL [reference formulation]), following intramuscular administration in patients with schizophrenia. The drug product—paliperidone palmitate 156 mg, hydrolyzes to paliperidone (the active moiety), resulting in a dose strength of paliperidone 100 mg. The secondary objective was to evaluate the safety and tolerability of the test formulation.
Methods
I. Study design
In this open-label, randomized, parallel, multiple-dose, multicentre study, a total of 267 adult patients with schizophrenia, who were on a steady once-monthly regimen of PP1M injectable suspension 156 mg administered intramuscularly, were enrolled. Patients from various parts of India were included in the study; all were of Asian ethnicity and identified as non-Hispanic and non-Latino. The study was conducted between January 2018 and January 2019.
The study was designed based on the FDA draft guidance for paliperidone palmitate (Office of Generic Drugs [OGD] Guidance on paliperidone palmitate, revised Jul 2016). The protocol, informed consent form, and all other relevant study documentation were reviewed and approved by the ethics committee. The study was conducted in accordance with the approved protocol, pertinent requirements of the Code of Federal Regulation (21 CFR), the Indian Council of Medical Research (ICMR) ethical guidelines, International Council for Harmonization (ICH) (Step 5) Guidance on Good Clinical Practice (E6), and Declaration of Helsinki. The study was executed by Cliantha Research Limited, a contract research organization, based in Ahmedabad, Gujarat, India.
a. Study phases
This study included 4 phases: (i) a screening period of 21 days prior to the administration of the first dose of the drug on Day 0 or the start of stabilization period (as applicable); (ii) stabilization period (if applicable) of at least 13 weeks, before randomization; (iii) treatment period of 24 weeks; and (iv) end of study treatment on Day 168 after randomization and safety follow-up of up to 28 days (± 4 days) after last sample collection.
b. Randomization
Subjects were randomized in a 1:1 ratio, after the stabilization period, to receive the test formulation or the reference formulation. In each treatment group, subjects were further randomized in 1:1 ratio based on the site of administration of the injection (gluteal or deltoid muscles).
c. Subject inclusion and exclusion criteria
Patients who met the following criteria were eligible to participate in the study – men or non-pregnant and non-lactating women, aged 18 to 65 years, clinically diagnosed with schizophrenia, on a stable monthly regimen of PP1M injectable suspension 156 mg (administered intramuscularly) prior to randomization, who have established tolerability to oral paliperidone, clinically stable and not hospitalized for exacerbation of psychiatric symptoms during 3 months prior to screening, with acceptable renal, haematological, and hepatic functions, and willing to provide written informed consent to participate in the study and abide by the study visit schedule.
Patients were excluded from the study if they had a history of known hypersensitivity to paliperidone, risperidone, or to any excipients in the treatments used in this study; dementia related psychosis; neuroleptic malignant syndrome while on treatment with atypical antipsychotics; concomitant use of medications known to prolong the QTc interval; uncontrolled metabolic disorders, cardiovascular disease, clinically significant hyperprolactinemia, severe hepatic impairment, tardive dyskinesia, seizures, idiopathic Parkinson’s disease, or cognitive and motor impairment; clinical global impression – severity of illness (CGI-S) score of 5 or more; on active treatment with drugs known to interact with paliperidone; on alcohol or drug-dependence during the 6-month period prior to screening; who attempted suicide within 12 months before screening or were at imminent risk of suicide; on treatment with risperidone long-acting injection, non-selective or irreversible monoamine oxidase inhibitor (MAOI), or electroconvulsive therapy before screening.
d. Treatment regimen
Stabilization period. The stabilization period was applicable to patients: (i) who were stable on oral paliperidone extended-release 9 mg regimen and were suitable candidates for paliperidone injectable therapy based on investigator assessment, or (ii) who had received less than 3 monthly-doses of PP1M injectable suspension 156 mg.
These patients were stabilized by administering at least 3 consecutive once-monthly maintenance doses of PP1M injectable suspension 156 mg (non-investigational product). The scheduled dosing days were Day 0, Day 7, Day 35, and Day 63 (± 2 days). An initiation dose of paliperidone palmitate extended-release injectable suspension 234 mg on Day 0 followed by 156 mg on days 7, 35, and 63, were administered intramuscularly. Patients who had received less than 3 monthly doses of PP1M injectable suspension 156 mg received the remaining doses during the stabilization period. Duration of stabilization period could be prolonged until the investigator determined that the patient was clinically stable.
Treatment period. Patients were administered PP1M injectable suspension 156 mg (either the test or reference formulation) at the deltoid or gluteal intramuscular injection site every 28 days (± 2 days) for a total of 6 doses, as per the randomization plan in a parallel design. The dosing schedule in the treatment period is detailed in Table 1.
Table 1: Dosing schedule during the treatment period
During the stabilization and treatment periods, patients were asked to be present at the study centre at least 2 hours before dosing to at least 4 hours post dosing. For dose 6 administration in the treatment period, subjects were housed in the clinical facility for at least 8 hours prior to dosing to at least 24 hours post-dose, after which patients visited the centre on ambulatory basis for pharmacokinetic blood sample collection. Patients were advised to fast for at least 4 hours prior to receiving the sixth dose and for at least 2 hours after receiving the sixth dose.
e. Blood sample collection
The blood sampling schedule was planned basis the known pharmacokinetics of paliperidone palmitate long-acting injection and standard pharmacokinetic principles. A total of 21 pharmacokinetic blood samples were collected per patient.
Pre-dose blood samples were collected 30 minutes prior to the administration of dose 4, 5, and 6 (on days 84, 112, and 140, respectively) and at the following times post-drug administration: (Day 140) 6, 12, 24 (Day 141), 36, 48 (Day 142), 60 (Day 142), 72 (Day 143), 96 (Day 144), 120 (Day 145), 144 (Day 146), 192 (Day 148), 240 (Day 150), 288 (Day 152), 336 (Day 154), 384 (Day 156), 480 (Day 160), 576 (Day 164) and 672 (Day 168) hours.
II. Analytical methods
The plasma samples (0.10 mL) were processed and analysed for paliperidone using validated analytical procedure by the Bioanalytical Laboratory of the Clinical Research Centre, Mylan Laboratories Ltd (Hyderabad, India). The procedure was validated for the quantitation of paliperidone in the range of 0.2 ng/mL to 100.218 ng/mL. The samples comprising the analytes of interest, paliperidone and the internal standard, D4-paliperidone, were processed using liquid-liquid extraction. The analytes were subjected to chromatography using an HPLC column prior to introducing to a mass spectrometer (Applied Biosystems API4000 LC/ MS/MS) set to operate in the positive ion selective reaction mode. Quantitation was performed using a weighted (1/x²) linear least squares regression analysis generated from calibration standards prepared immediately before each run.
III. Pharmacokinetic analysis
Multiple-dose (steady state) pharmacokinetic parameters for paliperidone after the administration of the 6th dose of either the test or reference formulation were determined using non-compartmental analysis.
- Area under the plasma concentration-time curve (AUCtau,ss) during the dosing interval at steady state
- The minimum concentration (Cmin,ss) at steady state following drug dosing, from the observed plasma concentration-time profile over the sampling time interval.
- The maximum concentration (Cmax,ss) at steady state and the time at which it occurred relative to the administered dose (Tmax,ss)
- The average concentration (Cav,ss) at steady state during a dosing interval, which is AUCtau/tau (where tau is the dosing interval)
- Fluctuation (%), which is the range of concentrations divided by the average steady-state concentration. Fluctuation (%) was calculated as [(Cmax,ss-Cmin,ss) / Cav,ss] × 100
IV. Statistical analysis
Statistical analyses were performed on the pharmacokinetic parameters using SAS Software (SAS Institute, Cary, NC). Steady state attainment was investigated separately for each formulation using the natural logarithm transformed pre-dose concentrations (CPD) for Dose 4 (Day 84), 5 (Day 112), and 6 (Day 140) to calculate the 95% CI upper bound of the slope. If the 95% CI upper bound of the slope was less than 0.2231 (natural log 1.25 = 0.2231), then it was concluded that the change in pre-dose concentrations was not significant, and that steady state was reached.
The primary assessment of bioequivalence was based on relative natural log transformed geometric least square mean ratio and the 90% confidence intervals for Cmax,ss and AUCtau,ss 90% CI being within 80.00% to 125.00% range.
Sample size estimation
The sample size estimation was carried out considering the following conditions for the primary PK analytes AUCtau,ss and Cmax,ss: T/R ratio = 95%; inter-subject CV (%) ~ 50%; significance level = 5%; power ≥80%; and bioequivalence limits = 80% to 125% along with an estimation for drop-out rate. Therefore, assuming a true ratio between 0.95 and 1.06 with an inter-subject variability of 44%, at least 78 subjects per treatment group were required to demonstrate bioequivalence with approximately 80% power. To accommodate potential withdrawals and dropouts due to AEs, non-compliance, or personal reasons (estimated at about 28%), a total of 106 subjects per treatment group need to be randomized and dosed.
V. Safety analysis
Demographic details, medical history (including psychiatric health) and current medical status, details about concomitant therapy or use of any medicine were recorded. Comprehensive physical examination, electrocardiogram (ECG), chest X-ray, and clinical laboratory investigations were conducted. Vitals signs (pulse rate, body temperature, respiratory rate, and blood pressure [especially to monitor orthostatic hypotension]), all adverse events (AEs), Clinical Global Impression (CGI) scale for severity of illness, body mass index (BMI), and injection site reaction were recorded and assessed at various time points of the study period.
Clinical laboratory investigations included haematology (including absolute neutrophil count, differential leukocyte count, and haemoglobin), liver enzymes, fasting and random blood sugar, HbA1c, lipid profile (serum cholesterol, triglyceride, low-density lipoprotein (LDL) and high-density lipoprotein (HDL) in fasting state), electrolytes (serum potassium and serum magnesium), total proteins, albumin, serum prolactin; serum pregnancy test; immunological tests (HIV (Human Immunodeficiency Virus), HBsAg (Hepatitis B surface antigen), Anti-HCV (Hepatitis C Virus)); urinalysis; alcohol breath test; and urine drug scan.
Results
I. Patient disposition
A total of 267 patients were enrolled and randomized, with 134 patients receiving the test formulation and 133 patients receiving the reference formulation. 253 patients successfully completed the study with 14 patients discontinued (4 from the test formulation group and 10 from the reference formulation group). Plasma samples from these 253 patients and one additional patient, who met the requirement of providing the required blood samples for pharmacokinetic analysis but discontinued later, (N = 254) were considered for the pharmacokinetic and statistical analysis. Patient disposition details are featured in Figure 1.
Figure 1: Study flow diagram summarizing patient disposition
II. Demographic Data
The demographic details of the study population (N = 254) considered for the pharmacokinetic analysis are featured in Table 2. Patients enrolled had a clinical diagnosis of schizophrenia as per the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-V) and CGI-S score of less than 5.
III. Pharmacokinetic and statistical results
a. Mean plasma concentrations at steady-state
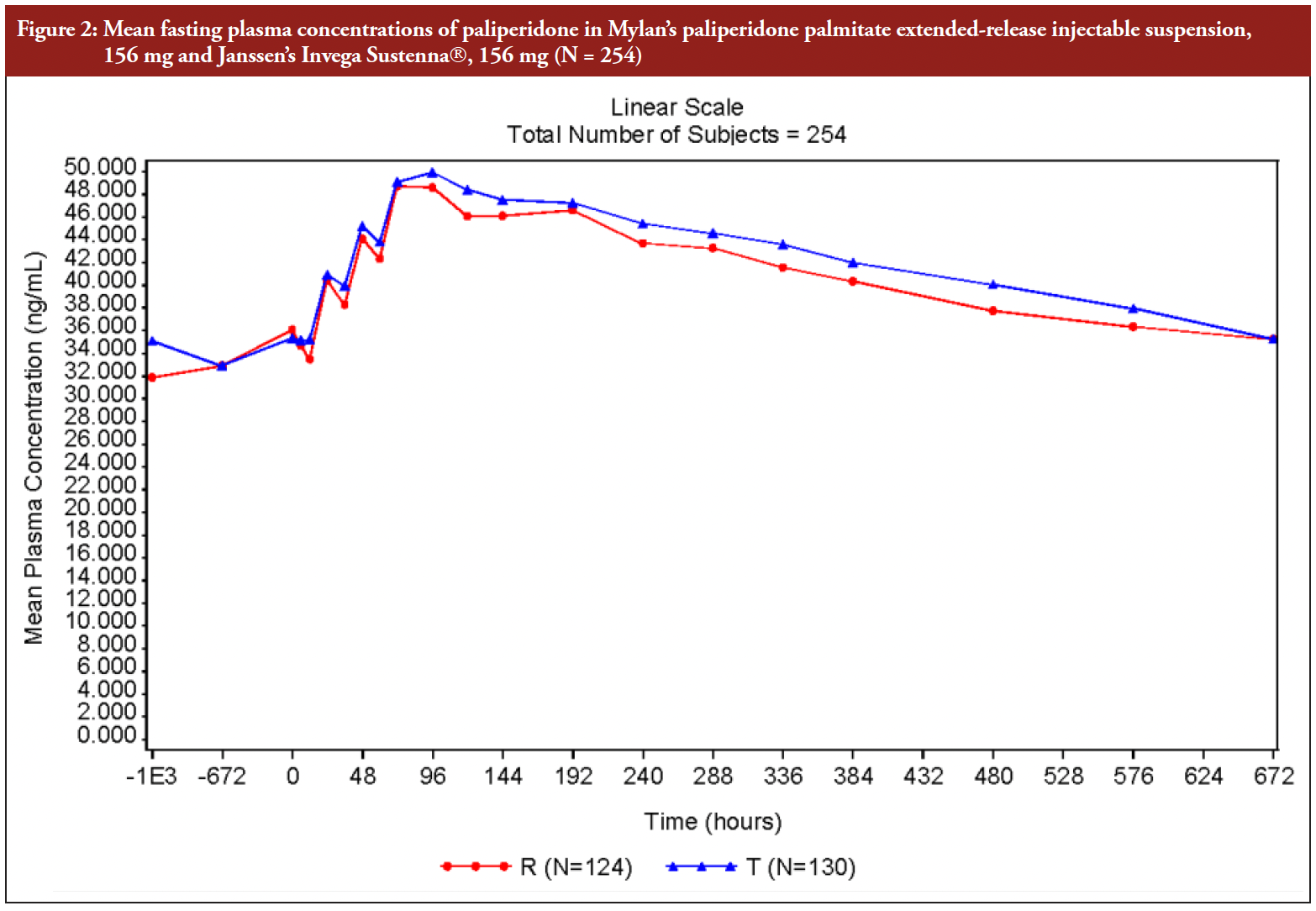
The mean plasma profiles analysed for paliperidone, at steady state, in the group administered with Mylan’s Paliperidone Palmitate Extended-release Injectable Suspension (156 mg) and the group administered with Janssen’s Invega Sustenna® (156 mg), were found to be similar.
The mean steady-state concentration of paliperidone versus time profile in both test and reference groups, is presented in Figure 2.
Figure 2: Mean fasting plasma concentrations of paliperidone in Mylan’s paliperidone palmitate extended-release injectable suspension, 156 mg and Janssen’s Invega Sustenna, 156 mg (N = 254)
b. Steady-state assessment
The attainment of steady state was assessed by examining the mean increase between the natural log transformed pre-dose concentrations (CPD) of Dose 4 (Day 84), Dose 5 (Day 112) and Dose 6 (Day 140). As shown in Table 3, the 95% CI upper bound of the slope (mean increase between doses) was 0.03821 and 0.08175 for test and reference product separately, that is less than 0.2231 (log1.25). Thus, it is concluded that steady state was achieved for both the test and reference formulations, see Table 3.
c. Bioequivalence assessment
The pharmacokinetic parameters for paliperidone, at steady state, were analysed using ANOVA. The geometric LS mean and arithmetic mean (%CV), LS means ratio, and the 90% CIs of the pharmacokinetic parameters analysed for paliperidone for Mylan’s PP1M 156 mg injectable suspension and Janssen’s Invega Sustenna® (PP1M 156 mg injectable suspension), are presented in Table 4.
The test and reference formulations demonstrated similar mean pharmacokinetic parameters and variability. The geometric mean ratio and the 90% CIs for the test to reference ratio for the natural log transformed parameters, AUCtau,ss and Cmax,ss, for paliperidone was within the 80.00% to 125.00% range. Thus, Mylan’s PP1M injectable suspension, 156 mg is bioequivalent to Janssen’s Invega Sustenna®, 156 mg following six once-monthly intramuscular dose administration to patients with schizophrenia.
IV. Safety results
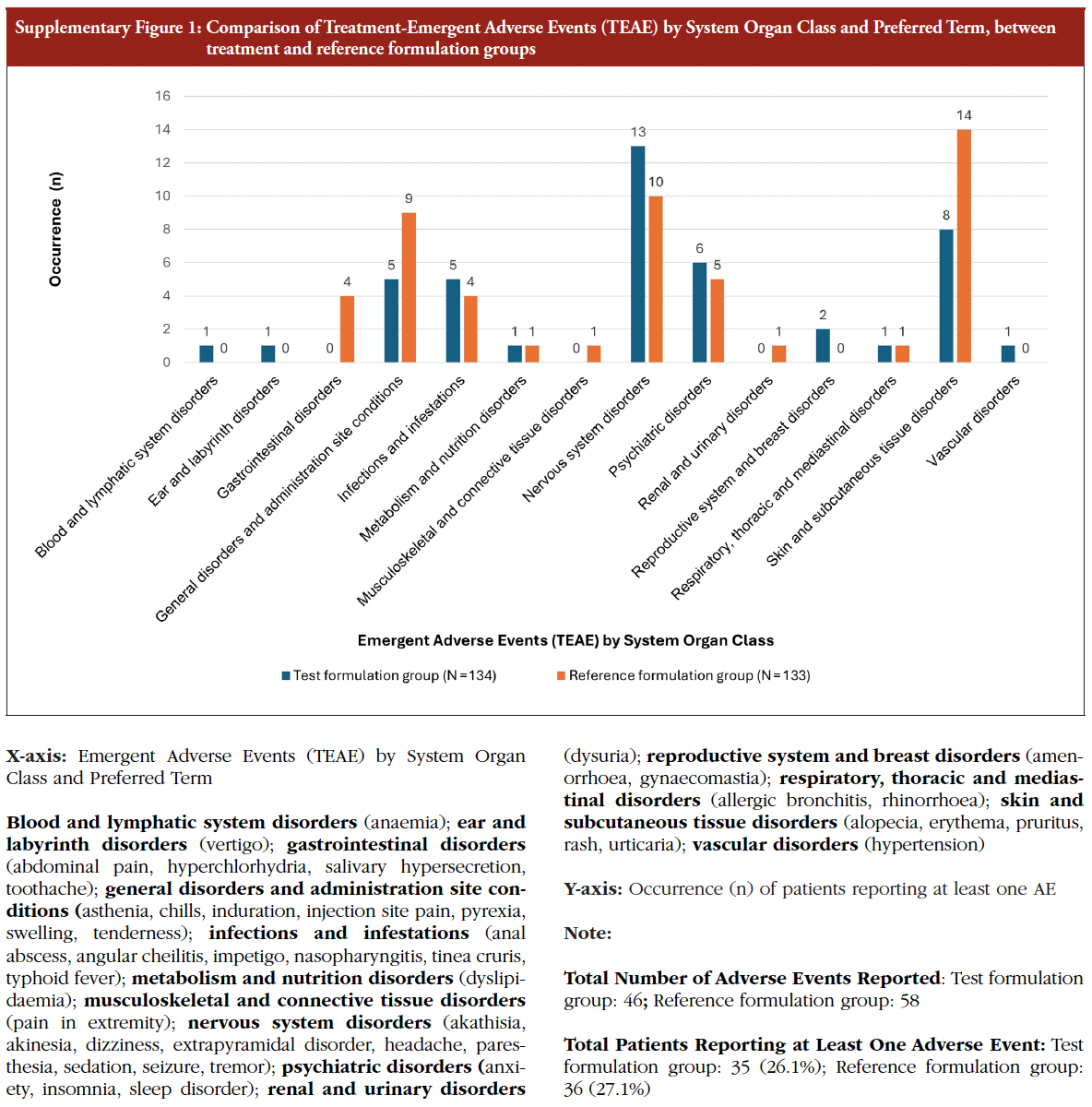
Patients were monitored for any adverse events (AEs) from the signing of the informed consent until completion of the study. No serious adverse events (SAEs) were reported during the treatment in randomized patients. A total of 46 treatment emergent adverse events (TEAEs) were reported by 35 (26.1%) patients during treatment with the test formulation and 58 TEAEs were reported by 36 (27.1%) patients during treatment with reference formulation. The percentage of patients who experienced an AE was comparable between the two treatment groups. Most of the AEs in both test and reference formulations were mild in intensity. There were no severe intensity AEs reported in either arm. Erythema was the most commonly reported AE in both groups, occurring in 6 out of 134 patients (4.5%) following the administration of the test formulation and in 10 out of 133 patients (7.5%) after administration of the reference formulation. One patient in the test group discontinued from the study due to adverse events – perianal abscess and anaemia, see Table 5. No notable differences in AEs based on the injection site were observed. A detailed break-up of the AEs reported in both treatment groups is presented in the Supplementary Figure 1.
Overall, PP1M injectable suspension was well tolerated following six once-monthly intramuscular injections of 156 mg (1×156 mg) dose administered to patients with schizophrenia.
Table 5: Adverse events reported in patients in the test and reference formulation groups
Discussion
The mechanism of action of paliperidone is unknown; however, it has been proposed that the drug’s therapeutic activity in schizophrenia is mediated through a combination of central dopamine Type 2 (D2) and serotonin Type 2 (5HT2A) receptor antagonism [13].
Following a single intramuscular dose, the plasma concentrations of paliperidone gradually rise to reach maximum plasma concentrations at a median Tmax of 13 days. The release of the drug starts as early as Day 1 and lasts for up to 126 days. The release profile and dosing regimen of paliperidone palmitate injectable suspension results in sustained therapeutic concentrations [13].
Although paliperidone is a strong D2-antagonist, which is believed to relieve the positive symptoms of schizophrenia, it causes less catalepsy and decreases motor functions less than traditional neuroleptics [14, 15]. Paliperidone LAI has a lower risk for negative outcomes compared to daily oral antipsychotics [16]. Also, paliperidone palmitate 1-month formulation is proven to be better in reducing the risk of relapse compared to oral paliperidone [17]. Published data on bioequivalence studies on LAIs is limited owing complex study-designs required by regulatory agencies and the corresponding methodological challenges. For instance, single-dose bioequivalence (BE) studies using the reference (typically highest) strength are generally recommended to differentiate formulations such as test and reference products. However, administering long-acting injectable (LAI) antipsychotics to healthy volunteers is unethical, so these studies are conducted in patients with schizophrenia [12, 18].
It is challenging to obtain a complete pharmacokinetic (PK) profile after a single dose because subsequent doses are administered at fixed intervals, requiring steady-state designs. While single-dose PK studies could be done in patients already stabilized on antipsychotics, practical and ethical concerns related to additive adverse events and recruitment difficulties limit this approach. Regulatory agencies differ in requirements: the European Medicines Agency (EMA) often requires both single-dose and multiple-dose steady-state studies for LAIs like paliperidone palmitate, while FDA primarily requires multiple-dose steady-state studies [18].
Conducting multiple-dose steady-state PK BE studies with LAIs in patients with schizophrenia presents unique challenges. The long half-life of LAI formulations precludes crossover designs, and high inter-subject variability in a parallel design necessitates large sample sizes and multi-centre recruitment of patients stably dosed with paliperidone. Strict control of dose timing is critical to avoid missed or delayed doses in this patient group, which is difficult over months across multiple sites.
Numerous blood samples (e.g. 21 in the current study) are required within a short timeframe to ensure steady state and PK profiling, demanding frequent patient visits. This is feasible in healthy volunteers at a single site but difficult in multi-centre patient studies. Additionally, variables affecting paliperidone pharmacokinetics such as injection site, gender, and BMI must be controlled, further complicating study conduct.
In this bioequivalence study, Mylan’s PP1M injectable suspension 156 mg (paliperidone palmitate 156 mg hydrolyzes to the active moiety, paliperidone, resulting in a dose strength of paliperidone 100 mg) was well tolerated following 6 monthly doses, intramuscularly (deltoid or gluteal). No serious adverse events (SAEs) were reported during the treatment in randomized subjects. The statistical analysis of the pharmacokinetic parameters showed that LSMeans ratio as well as the 90% CIs for the natural log transformed parameters, AUCtau and Cmax were within the prespecified bioequivalent range of 80% and 125% for paliperidone.
Conclusion
The study results demonstrate that Mylan’s paliperidone palmitate extended-release injectable suspension 156 mg is bioequivalent to Janssen Pharmaceuticals Inc’s Invega Sustenna® (paliperidone palmitate extended-release injectable suspension 156 mg), suggesting that the test formulation may be expected to deliver an equivalent therapeutic effect as its branded reference product. The availability of this complex generic with proven bioequivalence may provide clinicians with an additional treatment option for the management of schizophrenia and may improve access to patients.
Acknowledgement
The authors acknowledge all the principal investigators (Timir Shah, Vaishal Vora, Ratnakar Inamdar, Mahesh Chudgar, Rajendra Anand, Nehal Shah, K S Kulkarni, Nitin Dalaya, Amar Shinde, Sathyanarayana Rao, Radhika Reddy, Vipul Singh, Ramanathan Sathianathan, Bakul Buch, and Ramashanker Yadav) for their participation in the study.
The authors acknowledge Cliantha Research Limited, a contract research organization, based in Ahmedabad, Gujarat, India, involved in executing this trial. The authors also acknowledge the medical writing and editorial support provided by Shruthi VB, MSc, Dr Aswin Kumar Allupati, MBBS, and Dr Shantha Kumar V, PhD, from Viatris.
Funding sources
This study was funded by Viatris.
Author contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Ethics approval
All participants provided their written, informed consent to participate in the study and could withdraw their participation at any time. The study protocol, informed consent form, and all other relevant study documentation were reviewed and approved by the ethics committee. The study was conducted in accordance with the approved protocol, pertinent requirements of the Code of Federal Regulation (21 CFR), the Indian Council of Medical Research (ICMR) ethical guidelines, International Council for Harmonization (ICH) (Step 5) Guidance on Good Clinical Practice (E6), and Declaration of Helsinki.
Competing interests: OA is a paid consultant and speaker for pharmaceutical companies including Janssen-Ortho (Johnson & Johnson), Lundbeck, Otsuka, Boehringer Ingelheim, Allergan, AbbVie, Teva Pharmaceuticals, Newron Pharmaceuticals, HLS Therapeutics. JCG, MSL, PCG, MMT, and PP are employees of Viatris and JCG, MSL, and PP hold stocks of Viatris.
Periodic independent assessments were performed by external experts to ensure that the procedures followed and the data generated were in compliance with standard operating procedures, local and international regulatory requirements, ICH GCP and study requirements.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Ofer Agid1, MD
J Christopher Gorski2, PhD
Mark Shiyao Liu3, MS
Prasanna C Ganapathi3, MD
Mohna Mukund Toro3, MD
Pradeep Purushottamahanti4, MBBS, MBA
1Department of Psychiatry, Faculty of Medicine, University of
Toronto, Canada
2Global Clinical Pharmacology, Viatris Inc, USA
3Global Clinical Development, Viatris Inc, India
4Global Medical Affairs, Viatris Inc, India
References
1. Diagnostic and statistical manual of mental disorders, fifth edition, text revision (DSM-5-TR). American Psychiatric Association; 2022.
2. Agid O. Re-evaluating the prognosis of schizophrenia: tackling the issue of inadequate treatment. Expert Rev Neurother. 2024;24(9):831-5. doi:10.1080/1 4737175.2024.2365943
3. McCutcheon RA, Pillinger T, Varvari I, Halstead S, Ayinde OO, Crossley NA, et al. INTEGRATE: international guidelines for the algorithmic treatment of schizophrenia. Lancet Psychiatry. 2025;12(5):384-94. doi:10.1016/S2215-0366(25)00031-8
4. Velligan D, Salinas GD, Belcher E, Franzenberg KR, Suett M, Thompson S, et al. Clinician differences in attitudes and perceptions on the use of long-acting injectable antipsychotic agents in treating patients with schizophrenia: results from the US DECIDE survey. BMC Psychiatry. 2025;25(1):232. doi:10.1186/ s12888-025-06565-1
5. O‘Brien MN, Jiang W, Wang Y, Loffredo DM. Challenges and opportunities in the development of complex generic long-acting injectable drug products. J Control Release. 2021;336:144-58. doi:10.1016/j.jconrel.2021.06.017
6. Kishimoto T, Hagi K, Kurokawa S, Kane JM, Correll CU. Long-acting injectable versus oral antipsychotics for the maintenance treatment of schizophrenia: a systematic review and comparative meta-analysis of randomised, cohort, and pre-post studies. Lancet Psychiatry. 2021;8(5):387-404. doi:10.1016/ S2215-0366(21)00039-0
7. Haddad PM, Correll CU. Long-acting antipsychotics in the treatment of schizophrenia: opportunities and challenges. Expert Opin Pharmacother. 2023;24(4):473-93. doi:10.1080/14656566.2023.2181073
8. Vita G, Pollini D, Canozzi A, Papola D, Gastaldon C, Correll CU, et al. Efficacy and acceptability of long-acting antipsychotics in acutely ill individuals with schizophrenia-spectrum disorders: a systematic review and meta-analysis. Psychiatry Res. 2024:340:116124. doi:10.1016/j.psychres.2024.116124
9. Arango C, Fagiolini A, Gorwood P, Kane JM, Diaz-Mendoza S, Sahota N, et al. Delphi panel to obtain clinical consensus about using long-acting injectable antipsychotics to treat first-episode and early-phase schizophrenia: treatment goals and approaches to functional recovery. BMC Psychiatry. 2023;23(1):453. doi:10.1186/s12888-023-04928-0
10. Remington G, Addington D, Honer W, Ismail Z, Raedler T, Teehan M. Guidelines for the pharmacotherapy of schizophrenia in adults. Can J Psychiatry. 2017;62(9):604-16. doi:10.1177/0706743717720448
11. The American Psychiatric Association Practice Guideline for the Treatment of Patients With Schizophrenia, Third Edition. American Psychiatric Association. 2020.
12. U.S. Food and Drug Administration. Draft guidance on paliperidone palmitate. May 2023 [homepage on the Internet]. [cited 2025 Dec 1]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_022264.pdf
13. INVEGA SUSTENNA® (paliperidone palmitate) extended-release injectable suspension, for intramuscular use; [cited 2025 Dec 1]. Available from: https:// www.janssenlabels.com/package-insert/product-monograph/prescribing-information/INVEGA+SUSTENNA-pi.pdf
14. European Commission. Public Health – Union Register of medicinal products [homepage on the Internet]. [cited 2025 Dec 1]. Available from: https:// ec.europa.eu/health/documents/community-register/2019/20190516144963/ anx_144963_en.pdf
15. Shah S, Joshi D. Tolerability and efficacy of paliperidone ER compared to olanzapine in the treatment of schizophrenia: a randomized, double–blind, multicentric trial. Ind Psychiatry J. 2011;20(1):25-31. doi:10.4103/0972-6748.98411
16. Li P, Geng Z, Benson C, Patel C, Doshi JA. Real-world effectiveness of long-acting injectable and oral antipsychotic agents in US Medicare patients with schizophrenia. Adv Ther. 2025;42(2):1251-64. doi:10.1007/s12325-024-03075-6
17. Edinoff AN, Doppalapudi PK, Orellana C, Ochoa C, Patti S, Ghaffar Y. Paliperidone 3-month injection for treatment of schizophrenia: a narrative review. Front Psychiatry. 2021:12:699748. doi:10.3389/fpsyt.2021.699748
18. European Medicines Agency. Paliperidone palmitate depot suspension for injection 25 mg, 50 mg, 75 mg, 100 mg and 150 mg product specific bioequival-ence guidance [homepage on the Internet]. [cited 2025 Dec 1]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/paliperidone-palmitate-depot-suspension-injection-25-mg-50-mg-75-mg-100-mg-and- 150-mg-product-specific-bioequivalence-guidance_en.pdf
Author for correspondence: Pradeep Purushottamahanti, MBBS, MBA, Global Medical Affairs Viatris Inc, 7-11th Floor, Prestige Platina, Block 3 Kadubeesanahalli Village, Varthur Hobli, Outer Ring Road, Bengaluru 560103 Karnataka, India, email: pradeep.purushottamahanti@viatris.com |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2026 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



{kind=link}
{kind=link}