Differences in pharmacokinetic behaviour of branded enoxaparin and a US generic version in a non-human primate model
Published on 2014/12/16
Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(2):72-8.
Author byline as per print journal: Professor Walter P Jeske, PhD; Jeanine M Walenga, PhD; Nicolas Simon, MD, PhD; Debra Hoppensteadt, PhD; Josephine Cunanan, MD; Vicki Escalante, BS; Jawed Fareed, PhD; Mamdouh Bakhos, MD
|
Introduction: Low molecular weight heparins (LMWHs) are composed of a heterogeneous mixture of oligosaccharides that express a spectrum of biological activities. Studies conducted in primates (Macaca mulatta) comparing branded and one generic enoxaparin showed differences in pharmacodynamic (PD) behaviours, particularly in the mean release of tissue factor pathway inhibitor (TFPI). |
Submitted: 30 October 2014; Revised: 20 March 2015; Accepted: 21 March 2015; Published online first: 30 March 2015
Introduction
Low molecular weight heparins (LMWHs) are complex biological drug products derived from the chemical or enzymatic depolymerization of crude heparin [1]. This depolymerization yields a mixture of glycosaminoglycan chains of shorter length. While the composition of an LMWH product is dependent on the structural characteristics of the parent unfractionated heparin [2], some process-dependent structural modifications can also be induced [3, 4].
Approximately one quarter of the oligosaccharide chains in LMWH contain the pentasaccharide sequence required for potentiating the inhibition of factor Xa (FXa) and thrombin (factor IIa [FIIa]) (anti-FXa and anti-FIIa activities) by antithrombin [1, 5, 6]. Thus, the antithrombotic effects of the majority of the chains are not taken into account by these two anticoagulant activities. Yet these ‘forgotten’ chains are associated with the therapeutic effects of LMWH [7, 8].
Among the effects of the ‘forgotten’ glycosaminoglycans are interactions with the endothelial glycocalyx, which results in a modulation of the release of several factors such as myeloperoxidase [9], von Willebrand factor [10], and tissue factor pathway inhibitor (TFPI) [11, 12]. TPFI is a serine protease inhibitor that inactivates the TF/FVIIa complex that serves as the primary initiator of the coagulation cascade. TFPI is therefore a critically important naturally occurring inhibitor of thrombin generation that facilitates the antithrombotic activity of administered heparin.
Enoxaparin (Lovenox®/Clexane®) is a LMWH commonly used for the treatment and prevention of deep vein thrombosis and the treatment of the acute coronary syndrome. Today, several generic versions of this LMWH have been commercialized. Because LMWH is a biological product, with a highly heterogeneous structure and multiple modes of antithrombotic actions, how to define biosimilarity among LMWH preparations has been a source of controversy [13]. In the US, bioequivalence of generic LMWH preparations as compared to the branded/originator product has been defined by the US Food and Drug Administration (FDA) according to five criteria: equivalence of heparin source material and mode of depolymerization, equivalence of physicochemical properties, equivalence of disaccharide building blocks and sequence of oligosaccharide species, equivalence of biological/biochemical assays and equivalence of in vivo pharmacodynamic (PD) profile [14]. The currently approved generic enoxaparins, registered through an abbreviated generic pathway by FDA, did not require clinical studies.
Chemical methods to quantitate heparin levels are not available. Rather, heparin levels are determined using biological assays that measure a single effect. The pharmacokinetic (PK) behaviour of heparins and LMWHs are typically determined in terms of PD measures such as anti-FXa and anti-FIIa activity. In our previous investigations of enoxaparin using a non-human primate model [15], PD differences between the branded and a US generic version of enoxaparin were observed. Such differences were most notable in the extent of TFPI release. The objective of this study was to better explain and characterize the differences between branded and generic enoxaparin by applying a non-linear mixed effects model (NONMEM) to the previously collected data [16–19]. In this model, the ‘cause’ (anti-FXa, anti-FIIa) and ‘effect’ (TFPI release) characteristics of LMWHs over their residence time in circulation were analysed to determine if differences exist between branded and generic enoxaparin following single-dose administration.
Materials and methods
Test agents
Five batches of the branded enoxaparin Lovenox® (Sanofi US, Bridgewater, NJ) were compared to five batches of the first registered generic enoxaparin in the US (Sandoz US, Princeton, NJ).
Experimental system
Studies were conducted in a non-human primate model (Macaca mulatta) as previously described [15]. Primates were administered a single 1 mg/kg sc dose of enoxaparin. Treatment groups were comprised of three independent primates for each drug-batch. A two-week washout period was used between treatments.
Blood samples were collected at baseline and at 1, 3, 6, 12, and 24 hours post-drug administration. Plasma samples were prepared and stored frozen until they were evaluated in batches using established assays.
Anti-FXa activity was determined by an amidolytic assay as described previously [20] using human FXa (Enzyme Research Laboratories, South Bend, IN) and Spectrozyme Xa (American Diagnostica, Greenwich, CT), adapted to run on the ACL Elite analyzer (Instrumentation Laboratory, Bedford, MA).
Anti-FIIa activity was determined by an amidolytic assay as described previously [20] using human thrombin (Enzyme Research Laboratories, South Bend, IN) and Spectrozyme TH (American Diagnostica, Greenwich, CT), adapted to run on the ACL 300 Plus analyzer (Beckman Coulter, Fullerton, CA).
Plasma enoxaparin levels were determined by extrapolation from anti-FXa and anti-FIIa concentration response curves developed by supplementing each enoxaparin sample to pooled normal primate plasma over a concentration range of 0.1 to 2.5 μg/mL.
Total TFPI levels were determined by immunoassay (Asserachrom TFPI: Stago, Parsippany, NJ). TFPI levels were baseline corrected to minimize the effect of primate-to-primate differences in basal TPFI levels.
Anti-FXa and anti-FIIa modelling
Data were analysed using the non-linear mixed effect modelling software program NONMEM (Version 7.2, ICON Development Solutions, Baltimore, MD) with a Fortran compiler. The first order conditional estimation (FOCE) method was used for parameter estimation. Anti-FXa and anti-FII data were ascribed to an open one- or two-compartment model with zero or first order input. The decrease of the activity (output) was evaluated with a first order or a Michaelis-Menten equation. The inter-individual variability was modelled with an exponential model and the residual variability with additive, proportional or combined models. For evaluation of the goodness-of-fit, the following graphs were compared: observed versus predictions (DV-PRED), observed versus individual predictions (DV-IPRE), normalized predictive distribution errors versus time (npde-TIME), normalized predictive distribution errors versus predictions (npde-PRED) and observed versus time with 90% interval of prediction (visual predictive check). Diagnostic graphics and distribution statistics were obtained using the R program (R Foundation for Statistical Computing, Vienna, Austria). The influences of covariates (branded or generic product, body weight) on all parameters were then tested. The diagnostic plots described above, the change in objective function, and the change in parameter variability to selected factors that improved the model’s prediction were noted. A decrease of at least 6.61 in objective function value, i.e. Chi2 distribution with one degree of freedom for P < 0.01, relative to the base model was required for the addition of a single parameter to the model.
TFPI modelling
TFPI levels as a function of time and anti-FXa or anti-FIIa activity were ascribed to an indirect response model. In these models, enoxaparin was thought to stimulate the response production rate, Ktr*R0 (transit time rate constant multiplied by response at baseline). The model equation was then:

where R and STIM stood for the PD response and the stimulation linked with the anti-FXa or the anti-FIIa activity respectively. When the drug treatment started, the system was at steady state which is defined by the baseline parameter, R = R0.
The individual time-courses showed that there was a loss of drug effect during the treatment time. To take this into account, an empirical ability of releasing TFPI was defined as a function of time and drug exposure. The ability model equation was then:
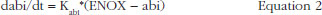
where Kabi, abi and ENOX stood for the transit time rate constant of ability, the ability of releasing TFPI over time and the effect of enoxaparin respectively.
The response model became:
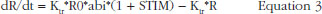
When anti-FXa or anti-FIIa activity increased, abi decreased and multiplied the response which decreased as well. STIM and ENOX took into account the enoxaparin exposure and were evaluated using linear, Emax and exponential models.
Anti-FXa or anti-FIIa and TFPI were analysed sequentially. A total of 15 animals with 144 observations of anti-FXa, 142 observations of anti-FIIa and 124 observations of TFPI were analysed. The anti-FXa or anti-FIIa parameters were fixed for the TFPI analysis. Additive and proportional models were tested to describe the residual and the inter-individual variability. The influence of the enoxaparin product (branded versus generic) was tested on each PD parameter. The criteria used for testing the effect of the enoxaparin version and for evaluating the goodness-of-fit were similar to those used for anti-FXa or anti-FIIa modelling. All modelling was performed using currently recognized techniques to describe the time course and effect of a test agent [21–23].
Results
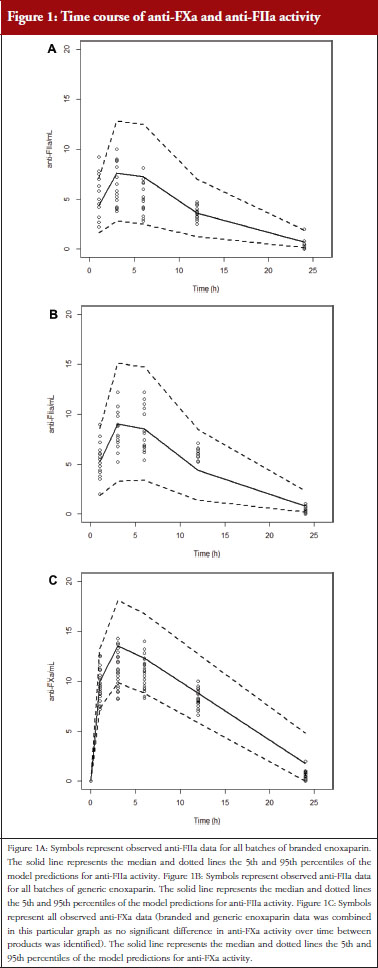
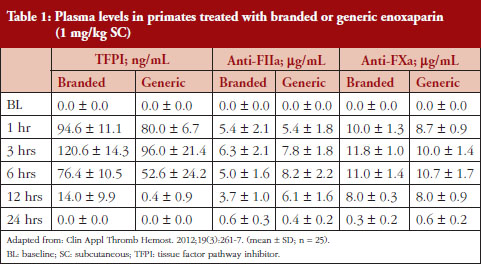
Administration of branded and generic enoxaparin has previously been shown to increase plasma TFPI concentration, as well as anti-FIIa and anti-FXa activities [15]. A higher peak TFPI concentration was observed following treatment with the branded enoxaparin compared to generic (P < 0.05), see Table 1. These higher TFPI levels were sustained through the 24-hour post-injection period. The anti-FIIa circulating activity over the 24-hour post-injection period was significantly higher for the generic, see Figure 1A, than the branded, see Figure 1B, enoxaparin (P < 0.05). The anti-FXa circulating activity, see Figure 1C, was not statistically different for the branded and generic enoxaparins.
Anti-FXa analysis
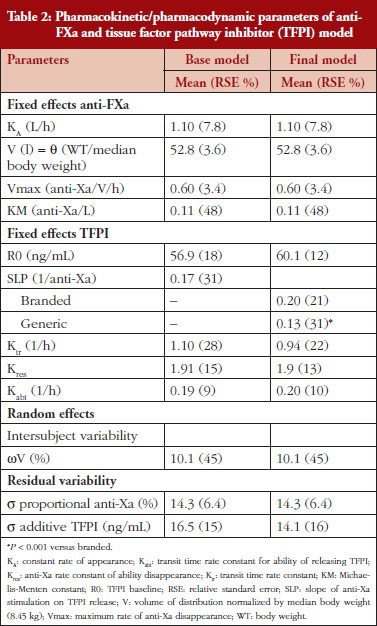
The anti-FXa activity versus time data were best described with a one-compartment model and a Michaelis-Menten elimination. The parameters were the constant of appearance (KA), the volume of distribution (V), the maximum rate of anti-FXa disappearance (Vmax), the Michaelis-Menten constant (KM), the inter-individual variability on the volume of distribution and the residual variability described as a proportional model. The only significant covariate that influenced the model was body weight (WT) on the volume of distribution. The circulating anti-FXa activity was not different between the branded and generic products, see Table 1. The final parameters are described in Table 2; all parameters were well estimated according to their relative standard errors (RSE). Figure 1C depicts anti-FXa observed time-course with the median and 5th/95th percentiles of the model predictions. The observed data are well included in the simulated percentiles with no significant bias.
Anti-FIIa analysis
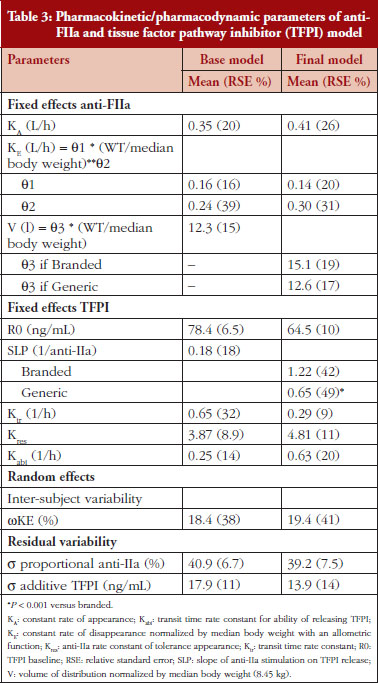
The anti-FIIa activity versus time data were best described with a one-compartment model and a first-order elimination. The parameters were the constant of appearance (KA), the volume of distribution (V), the constant of disappearance (KE), the inter-individual variability on the KE and the residual variability described as a proportional model. The WT was incorporated in the model for KE and V as an allometric function. The modelling analysis shows that the covariate, branded or generic enoxaparin, leads to a significantly different volume of distribution, see Table 3. The time-course of anti-IIa activity is different for the generic relative to the branded product. The final parameters are described in Table 3; all parameters were well estimated according to RSE. Figures 1A and 1B depict anti-FIIa observed time-course with the median and 5th/95th percentiles of the model predictions. The observed data are well included in the simulated percentiles with no significant bias.
Anti-FXa/TFPI analysis
Administration of both branded and generic enoxaparin was associated with an increase in TFPI concentration.
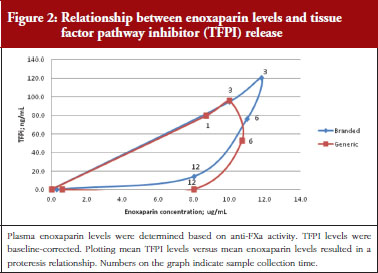
The exploratory plot of TFPI release versus anti-FXa activity as a measure of enoxaparin circulating concentration, see Figure 2, showed a clockwise hysteresis (proteresis) which was overlapping for branded and generic enoxaparin. Because the proteresis could not be fully described by an indirect response model, component taking into account the limited ability of releasing TFPI was added in the model. The capacity of enoxaparin to stimulate the release of TFPI (STIM defined in Eq. 1; SLP defined as the slope of anti-FXa stimulation on TFPI release) was defined as proportional because an Emax model was over-parameterized.

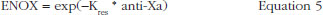
The effect of enoxaparin on the ability disappearance of releasing TFPI was estimated by the use of an exponential function (ENOX in Eq. 2) where Kres is defined as the anti-FXa rate constant of ability disappearance:
The final model is depicted in Figure 3. The parameters were the TFPI baseline (R0), the slope of anti-FXa stimulation on TFPI release (SLP), the transit time rate constant of TFPI release (Ktr), the transit time constant of ability of TFPI release (Kabi), and the anti-FXa rate constant on ability of TFPI release. The residual variability was better described as proportional but the inter-individual variability could not be correctly estimated. Three enoxaparin doses have been simulated (500, 1,000, 1,500 IU) to depict TFPI release and the decreasing ability to release TFPI, see Figure 4. With time the ability of releasing TFPI returns to the base value.
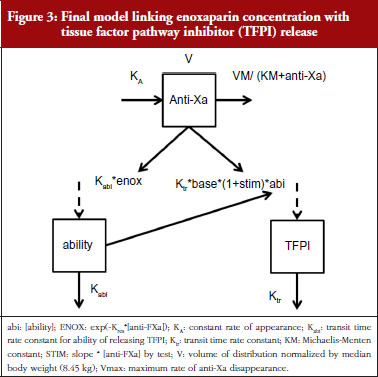
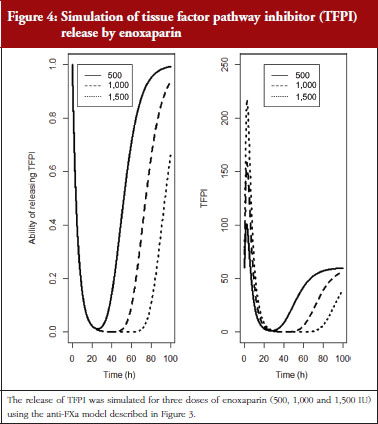
In the primate model, the TFPI values were different between the branded and generic products. This effect was significantly described by SLP values for branded and generic enoxaparin (P < 0.001). The parameters using the final model are described in Table 2.
Anti-FIIa/TFPI analysis
Administration of both branded and generic enoxaparin was associated with an increase in TFPI concentration.
The exploratory plot of TFPI release versus anti-FIIa activity as a measure of enoxaparin circulating concentration, see Figure 5, showed a clockwise hysteresis (proteresis) with distinct loops obtained for branded and generic enoxaparins. The proteresis could not be fully described by an indirect response model. Thus, as for the anti-FXa activity/TFPI release proteresis relationship, a component taking into account the ability to release TFPI was added in the model. The stimulation of enoxaparin on TFPI release (STIM) and the effect of enoxaparin on the ability disappearance of releasing TFPI (ENOX) were best estimated using similar functions to those previously defined for anti-FXa activity (Eq. 4 and 5).
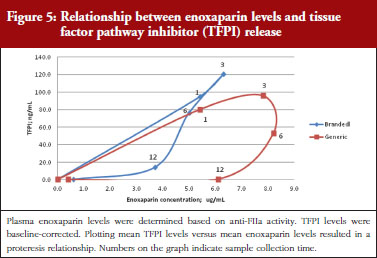
The parameters were the TFPI baseline (R0), the slope of anti-FIIa stimulation on TFPI release (SLP), the transit time rate constant of TFPI release (Ktr), the transit time constant of ability of TFPI release (Kabi), the anti-FIIa rate constant on ability of TFPI release. The residual variability was better described as proportional but the inter-individual variability could not be correctly estimated.
Data from the primate model demonstrated that the TFPI values were different between the branded and generic products. This effect was significantly described by the SLP values for branded and generic enoxaparin (P < 0.001). The parameters using the final model are described in Table 3.
Discussion
LMWH is not a single entity. Rather, it is a complex drug composed of a mixture of oligosaccharide chains that are heterogeneous in terms of their chemistry and biology. Heparin oligosaccharides vary in molecular weight (chain length) as well as in fine chemical structure, e.g. degree and pattern of sulfation. It is the combination of these chemical properties of heparin that govern its biological activities. As each heparin chain can be characterized by its individual chemical properties, each heparin chain will possess its individual biological effects.
Evaluation of heparins by in vitro analysis (essentially 100% bioavailability of all saccharides) will assess its activities as a whole, with the notable exclusion of those actions induced physiologically with administration of the heparin (such as TFPI release). Conversely, an in vivo/ex vivo analysis takes into account all biological activities. For heparins, inclusion of the PK behaviour is highly important because of the heterogeneous oligosaccharide chains that are variably absorbed/eliminated with subcutaneous administration. Thus, PD studies of a LMWH are the most reflective of how these drugs will behave in treated patients (true PK studies of heparin are not possible due to the heterogeneity of the drug’s molecular composition). PK behaviour is influenced by chain length with shorter heparin chains exhibiting a higher bioavailability and slower clearance than larger chains [5]. In terms of PDs, it is known that there is a minimal heparin chain length required to promote thrombin inhibition [24]. Thus, the PK behaviour of the individual heparin chains directly impacts the resulting biological activities.
It is widely accepted that heparin’s anticoagulant activity is largely (but not solely) derived through its ability to bind to antithrombin and accelerate its inhibition of a variety of coagulation factors. Since the specific pentasaccharide sequence required for high-affinity binding to antithrombin is present in only a small fraction of heparin oligosaccharides, measuring the inhibition of FXa or thrombin alone likely does not represent a full picture of heparin’s biological activity. This is supported by the fact that animal and human studies have not convincingly demonstrated a correlation between circulating anti-FXa activity and antithrombotic or haemorrhagic activity of LMWHs [25].
Another important mediator of the antithrombotic effect induced by heparin is TFPI release. TFPI is normally found in the vascular endothelium and is released into circulation following the administration of a polyanion such as heparin [12]. Previous studies have shown that the size and charge density of the polyanion influence the extent of TFPI release [12, 26], with larger, more highly charged oligosaccharides having a larger effect on TFPI release.
Our assessment of the PD of branded/originator versus generic enoxaparin used an integrated approach pairing known antithrombotic activities of LMWH over the residence time of the drug, i.e. drug level (measured by anti-FXa or anti-FIIa activity) paired with biological effect (TFPI release). These parameters (anti-FXa, anti-FIIa and TFPI) are among those recommended for pharmacological characterization of innovator and biosimilar LMWHs [27]. A previous study in healthy male volunteers receiving an infusion of unfractionated heparin demonstrated a clockwise proteresis loop when the anti-FIIa activity was plotted against TFPI levels [28]. This proteresis was believed to be due to an acute tolerance of endothelial TFPI release. This was also observed in our study with LMWH performed in non-human primates.
Importantly, our study revealed that there are PD differences in TFPI release between branded and generic enoxaparins. We suggest that this is due to compositional differences in the oligosaccharide components of the different enoxaparins (size and/or charge density of the saccharide chains) resident in blood circulation at a given time. The higher molecular weight chains would be particularly impactful in this regard. Heparin chains of varying molecular weight exhibit varying pharmacologic effects [28–30] with TFPI release being more dependent on the higher molecular weight chains [12, 26].
The findings of our study, based on ex vivo analyses, demonstrated that despite the similarity of the resulting anti-FXa activity for the branded/originator and generic enoxaparins following subcutaneous administration, there are significant differences between these two enoxaparin products in terms of anti-FIIa activity and other heparin-dependent biological activities. The higher variability in the TFPI release for the generic enoxaparin, and the difference in the TFPI release (in total and over time) between the two enoxaparin products, suggests the possibility that the higher molecular weight saccharide chains are structurally different between branded and generic enoxaparins. This may be a result of subtle differences in the depolymerization process or in the composition of the parent unfractionated heparin. Results from our other investigations on the same batches of these enoxaparins revealed a higher biological variability among the batches of the generic enoxaparin in blood coagulation assays where activity is based on the higher molecular weight components of the heparin. This further supports the concept that differences between the branded/originator and generic enoxaparin products exist in the higher molecular weight components [15, 31].
Our study does have some limitations. A crossover design was not applied. Additionally, there was insufficient data to be able to test for differences between drug batches.
The data of this study and its analytical outcome have a two-fold implication on LMWH pharmaceutical products. First, product characterization to determine equivalence to the originator product, and thus clinical interchangeability of a generic LMWH, is more complex than simple anti-FXa and anti-FIIa measurements and the required chemical characterization that focuses on the lowest molecular weight oligosaccharides and disaccharides. Second, the demonstrated differences between the branded/originator and generic enoxaparin products in terms of temporal behaviour on important antithrombotic properties may have implications on the therapeutic index of LMWHs, particularly in clinical settings where higher dosages are administered such as in treatment of acute coronary syndrome.
Disclosure of financial and competing interests: This study was supported by a research grant to Dr Jeanine M Walenga from Sanofi. The sponsor, Sanofi, had no input on the study design or data analysis.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Professor Walter P Jeske1,2, PhD
Jeanine M Walenga1,2, PhD
Nicolas Simon3, MD, PhD
Debra Hoppensteadt2, PhD
Josephine Cunanan2, MD
Vicki Escalante1, BS
Jawed Fareed2, PhD
Mamdouh Bakhos1, MD
1Department Thoracic and Cardiovascular Surgery, Loyola University Chicago, Stritch School of Medicine, Health Sciences Division, Room 5210, Building 110, 2160 S First Avenue, Maywood, IL 60153, USA
2Department of Pathology, Stritch School of Medicine, Loyola University Chicago, 2160 S First Avenue, Maywood, IL 60153, USA
3Aix-Marseille Université, AP-HM, Hôpital Sainte Marguerite, Service de Pharmacologie Clinique, FR-13274 Marseille, France
References
1. Weitz JI. Low-molecular-weight heparins. N Engl J Med. 1997;337(10):688-98.
2. Bianchini P, Mascellani G. Few bicyclic acetals at reducing end of low-molecular-weight heparins: might they restrict specification of pharmacopoeia? Pharmeur Sci Notes. 2005(1):1-3.
3. Linhardt RJ, Gunay NS. Production and chemical processing of low molecular weight heparins. Semin Thromb Hemost. 1999;25(Suppl 3):5-16.
4. Ozug J, Wudyka S, Gunay NS, et al. Structural elucidation of the tetrasaccharide pool in enoxaparin sodium. Anal Bioanal Chem. 2012;403:2733-44.
5. Hirsh J, Raschke R. Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126(3 Suppl):188S-203S.
6. Viskov C, Just M, Laux V, Mourier P, Lorenz M. Description of the chemical and pharmacological characteristics of a new hemisynthetic ultra-low-molecular-weight heparin, AVE5026. J Thromb Haemost. 2009;7(7):1143-51.
7. Fareed J, Jeske W, Hoppensteadt D, Clarizio R, Walenga JM. Are the available low-molecular-weight heparin preparations the same? Semin Thromb Hemost. 1996;22 Suppl 1):77-91.
8. Drouet L. Low molecular weight heparin biosimilars: how much similarity for how much clinical benefit? Target Oncol. 2012;7 Suppl 1:S35-42.
9. Baldus S, Rudolph V, Roiss M, et al. Heparins increase endothelial nitric oxide bioavailability by liberating vessel-immobilized myeloperoxidase. Circulation. 2006;113(15):1871-8.
10. Montalescot G, Bal-dit-Sollier C, Chibedi D, et al. Comparison of effects on markers of blood cell activation of enoxaparin, dalteparin, and unfractionated heparin in patients with unstable angina pectoris or non-ST-segment elevation acute myocardial infarction (the ARMADA study). Am J Cardiol. 2003;91(8):925-30.
11. Hansen JB, Sandset PM, Huseby KR, et al. Differential effect of unfractionated heparin and low molecular weight heparin on intravascular tissue factor pathway inhibitor: evidence for a difference in antithrombotic action. Br J Haematol. 1998;101(4):638-46.
12. Ma Q, Tobu M, Schultz C, et al. Molecular weight dependent tissue factor pathway inhibitor release by heparin and heparin oligosaccharides. Thromb Res. 2007;119(5):653-61.
13. Harenberg J, Kakkar A, Bergqvist D, et al. Recommendations on biosimilar low-molecular-weight heparins. J Thromb Haemost. 2009;7(7):1222-5.
14. U.S. Food and Drug Administration. Establishing active ingredient sameness for a generic enoxaparin sodium, a low molecular weight heparin [homepage on the Internet]. [cited 2015 Mar 20]. Available from: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm220023.htm
15. Walenga JM, Jeske WP, Hoppensteadt D, et al. Comparative studies on branded enoxaparin and a US generic version of enoxaparin. Clin Appl Thromb Hemost. 2013;19(3):261-7.
16. Danhof M, de Lange EC, Della Pasqua OE, Ploeger BA, Voskuyl RA. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol Sci. 2008;29(4):186-91.
17. Kwon Y. Handbook of essential pharmacokinetics, pharmacodynamics and drug metabolism for industrial scientists. Springer; 2001. p. 203-4.
18. Toutain PL. Pharmacokinetic/pharmacodynamic integration in drug development and dosage-regimen optimization for veterinary medicine. AAPS Pharm Sci. 2002;4(4):1-29.
19. Brindley CJ, Taylor T, Diness V, Oestergaard PB, Chasseaud LF. Relationship between pharmacokinetics and pharmacodynamics of tinzaparin (logiparin), a low molecular weight heparin, in dogs. Xenobiotica.1993;23(6):575-88.
20. Walenga JM, Fareed J, Messmore HL Jr. Newer avenues in the monitoring of antithrombotic therapy: the role of automation. Semin Thromb Hemost. 1983;9(4):346-54.
21. Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development. CPT Pharmacometrics Syst Pharmacol. 2012;1:e6. doi:10.1038/psp.2012.4
22. Mould DR, Upton RN. Basic concepts in population modeling, simulation and model-based drug development-part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst Pharmacol. 2013;2:e38. doi:10.1038/psp.2013.14
23. U.S. Food and Drug Administration. Guidance for industry: population pharmacokinetics. February 1999 [homepage on the Internet]. 2010 Feb 10 [cited 2015 Mar 20]. Available from: http://www.fda.gov/downloads/Drugs/Guidances/UCM072137.pdf
24. Bray B, Lane DA, Freyssinet JM, Pejler G, Lindahl U. Anti-thrombin activities of heparin. Effect of saccharide chain length on thrombin inhibition by heparin cofactor II and by antithrombin. Biochem J. 1989;262(1):225-32.
25. Samama MM, Gerotziafas GT. Comparative pharmacokinetics of LMWHs. Semin Thromb Hemost. 2000;26 Suppl 1:31-8.
26. Alban S. Molecular weight-dependent influence of heparin on the form of tissue factor pathway inhibitor circulating in plasma. Semin Thromb Hemost. 2001;27(5):503-11.
27. Harenberg J, Walenga J, Torri G, Dahl OE, Drouet L, Fareed J. Update of the recommendations on biosimilar low-molecular weight heparins from the Scientific Subcommittee on Control of Anticoagulation of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2013;11(7):1421-5.
28. Kemme MJ, Burggraaf J, Schoemaker RC, Kluft C, Cohen AF. Quantification of heparin-induced TFPI release: a maximum release at low heparin dose. Br J Clin Pharmacol. 2002;54(6):627-34.
29. Holmer E, Kurachi K, Soderstrom G. The molecular-weight dependence of the rate-enhancing effect of heparin on the inhibition of thrombin, factor Xa, factor IXa, factor XIa, factor XIIa and kallikrein by antithrombin. Biochem J. 1981;193(2):395-400.
30. Khorana AA, Sahni A, Altland OD, Francis CW. Heparin inhibition of endothelial cell proliferation and organization is dependent on molecular weight. Arterioscler Thromb Vasc Biol. 2003;23:2110-15.
31. Walenga JM, Jeske WP, Escalante V, Hoppensteadt D, Fareed J, Bakhos M. Thromboelastographic evaluation of blood coagulation in the presence of branded and generic enoxaparins. Int Angiol. 2012;31(6):517-25.
|
Author for correspondence: Professor Walter P Jeske, PhD, Professor, Thoracic and Cardiovascular Surgery, Loyola University Chicago, Stritch School of Medicine, Health Sciences Division, Room 5210, Building 110, 2160 S First Avenue, Maywood, IL 60153, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



Journal of Pharmacokinetics & Experimental Therapeutics is an open access journal that focuses on pharmacokinetic properties of drugs, including Absorption, Distribution, Metabolism and Excretion (ADME) while underlining the importance of experimental therapeutics in pharmacokinetic research.
Journal of Pharmacokinetics & Experimental Therapeutics accepts manuscripts in clinical pharmacokinetics, population pharmacokinetics, antibody pharmacokinetics, pharmacokinetics review, vancomycin pharmacokinetics, tacrolimus pharmacokinetics, pharmacokinetic drugs, pharmacokinetics intravenous, pharmacokinetics oral, pharmacokinetics renal, pharmacokinetics human, pharmacokinetics rat, pharmacokinetics mice, pharmacokinetics for children, pharmacokinetics research to promote healthy life are welcome.