Interchangeability. An insurmountable fifth hurdle?
Published on 2014/04/23
Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):88-93.
|
Abstract: |
Submitted: 24 February 2014; Revised: 31 March 2014; Accepted: 3 April 2014; Published online first: 16 April 2014
Introduction
In September 2013, the first biosimilar version of a monoclonal antibody (mAb) was authorized in the European Union (EU). The availability of lower-priced, therapeutically equivalent alternatives will make prescribing driven more by market forces, especially in those countries where patients pay out-of-pocket, but also for evermore constraint healthcare systems. Biotechnology product sales have grown from US$36 billion in 2002 to US$163 billion in 2012 and there will be increasing pressure on healthcare systems to reduce drug expenditures [1]. The promise of cost savings is the main potential of biosimilars. The price reductions for currently authorized biosimilars have been modest (20–30%), it is expected that the price reductions for biosimilar monoclonal antibodies will be larger (up to 50%) [2, 3]. Key to the commercial success of biosimilars is the way they are accepted for use in clinical practice and if and when patients can be switched to and from innovator products and biosimilars [4].
Because confusion about the distinct notions of switching, interchangeability and substitutability have tended to confound the discussion, a recent European Commission sponsored consensus document provides the definitions of these terms, as shown in Table 1.

The EU regulatory designation of biosimilarity is without prejudice to prescribing and reimbursement decisions, which remain the jurisdiction of individual EU Member States. However, EU regulators have asserted that biosimilar products that are approved according to EU regulatory standards should be considered as interchangeable with the reference product, at least from the perspective of being therapeutically equivalent alternatives [5]. Thus, the EU ‘biosimilar’ status should represent sufficient assurance for prescribing physicians to use biosimilar medicines interchangeably with the originator product. However, this does not mean that pharmacists would be able to substitute automatically one product version for another – since there is no implication of interchangeable status for substitutability within EU Pharmaceutical Law governing Similar Biological Medicinal Products.
The regulatory position is different in the US, because the US Food and Drug Administration (FDA) does have jurisdiction to classify biosimilar products as ‘interchangeable’, and this designation then enables substitution at the pharmacy level without the consent of the prescribing physician – provided that state legislature permits such substitution and that the prescribing physician has not indicated in the prescription form that the prescribed product must not be substituted. The US legal provisions for biosimilar medicinal products define additional criteria for designation of a biosimilar product as interchangeable, as described in Table 2.

Here we will discuss challenges to the regulatory approach to establishing interchangeability, in the sense of considering biosimilar versions as therapeutic equivalents that could – depending on National or Federal Law – be substituted at the pharmacy level, and compare it to the weight of real-world evidence of the risks of potential differences between two monoclonal antibodies [6]. We will centre most of our discussion on tumour necrosis factor-alpha inhibitors (TNFIs).
Challenges to establish interchangeability
The US legal pathway for biosimilars does not define the weight of evidence required to fulfil the requirement that an interchangeable biosimilar product ‘can be expected to produce the same clinical result as the reference product in any given patient’ and ‘the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product without such alternation or switch’ [7].
The EU regulatory process evaluates biosimilarity, i.e. similarity in direct comparison to the reference product, at a fixed point in time, namely the point in time when Marketing Authorization is granted. Although formal designation of interchangeability is not a feature of the EU regulatory process for biosimilars, the closely perceived relationship of interchangeability to switchability – effectively, regulatory approval of a medicinal product as ‘biosimilar’ could and should be regarded by physicians as a clear signal that patients may safely be switched between therapeutic alternative for economic reasons, on the basis of rigorous regulatory standards – implies a need to reflect on the weight of evidence required for sound clinical decision making.
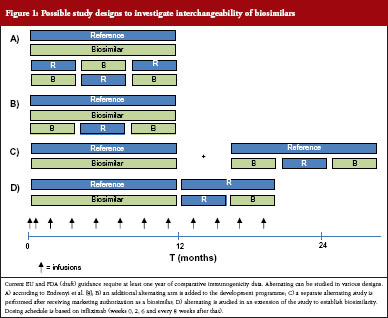
What would a study investigating interchangeability of biosimilars look like? For small molecule generics, investigation of the clinical impact of interchangeability is not part of the preauthorization regulatory requirements, see table 3. Endrenyi et al. state that, to establish comparable efficacy and safety of alternating, at least two switches are required [8]. Current EU and (draft) FDA guidance require at least one year of comparative clinical data for products that are administered chronically [9, 10]. Therefore, to establish interchangeability, a parallel arm could be included which includes at least two switches. Several designs have been suggested and it has been proposed to include a combined RTR, TRT design, where R is the reference product and T the biosimilar product, see Figure 1A [8]. However, it may not be necessary to include two alternating arms, see Figure 1B. Alternatively, switching and/or alternating can be studied in a supplementary study or as an extension to the registration study, see Figure 1C and Figure 1D. Such a design could establish that a population that is alternated has comparable efficacy and safety to a non-alternated group. A question that remains is, if a product is deemed interchangeable with the reference product, does this automatically also mean that a product is interchangeable with other (interchangeable) biosimilars?

What to measure, who to measure and for how long?
Interchanging would likely take place mostly in patients that are (at least initially) responding to the treatment. Therefore, it may be necessary to include only responders in a study investigating interchangeability. A design that includes a run-in phase to select only the responders to induction therapy would be a possibility, and has been applied to studying TNFIs in Crohn’s disease [11]. Studies including crossovers have been performed for epoetin biosimilars authorized in the EU, but these did not compare the switched arm with reference product monotherapy [12]. For epoetins this is feasible as easily measurable pharmacodynamic markers are available to assess the product’s efficacy, whereas for monoclonal antibodies such markers rarely exist. If such a design were chosen, the next questions would be: (i) what duration of evaluation is required following switching; and (ii) what response margin should be predefined for establishing sustainability of efficacy? Patients would need to be evaluated following the switch for a period that would allow detection of a clinically relevant difference. Data from infliximab shows that the mean time to relapse after discontinuing infliximab in rheumatoid arthritis patients varies from 15 weeks to 17 months [13]. The median time to relapse in Crohn’s disease patients with a sustained response was 16.4 months after withdrawing infliximab and 43.9% of patients relapsed within one year [14]. These studies investigating withdrawal focused on patients with sustained response to infliximab. Nevertheless, a very large study may be required to gain enough power to be able to actually establish an efficacy difference at a predefined, clinically relevant margin. Also, anti-drug antibodies (ADA) will need to be determined at baseline and throughout the study. Perhaps, a more sensitive approach would be to measure product trough levels as a surrogate for the impact of the ADA on efficacy. The impact of detected ADA’s would depend on impact on drug levels relative to dose–response relationship in the target population. This would require that this relationship has been well defined for the reference product – which is not always the case.
In the absence of any biosimilar product approvals in the US, it is not surprising that FDA has hesitated to provide more specific requirements for meeting the regulatory standard for designation of interchangeability. The most likely scenario is for interchangeability to be considered at a second stage, following initial approval as a ‘non-interchangeable’ biosimilar. Otherwise, initial regulatory approval, as well as uptake into clinical practice, could be hindered by uncertainties about the standards required for designation of interchangeability.
Residual safety concerns at time of authorization that may prevent interchanging
In order to be authorized, a biosimilar mAb must demonstrate comparable efficacy and safety to the reference product in a sensitive clinical model. Pre-authorization clinical trials are usually underpowered to assess possible differences in safety events between two products. So, if different authorized products remain within the defined specification limits (that are aimed to fall within the observed values of the originator) what kind of adverse events may be expected to occur as a result of interchanging biosimilar products? Limited data are available, but the experience accumulated to date does not indicate that there is a safety risk of switching between therapeutic alternative biotherapeutic products or biosimilars [15, 16].
A potential concern for biosimilars is the possibility of differences, relative to the reference product, in the dynamics of formation of clinically relevant ADA that could influence overall benefit-to-risk. Undesirable immunogenicity has been identified as factor to be assessed for the registration of therapeutic monoclonal antibodies, including biosimilars, because this has been linked to reduced efficacy, infusion reactions and hypersensitivity reactions. The formation of ADA associated immune complexes has also been associated with an increase in thromboembolic events in patients treated with adalimumab [17]. It has been questioned whether interchanging a biological therapy with a biosimilar agent ‘might promote increased immunogenicity that could compromise the efficacy and safety of both medications’ [18]. However, there is no evidence that (repeated) switching per se induces immunogenicity [15]. Most antibodies targeting infliximab target the murine part, whereas antibodies targeting adalimumab are aimed at the complementarity-determining region [17]. Biosimilar products meeting the EU and US regulatory standards are required to have a fully identical amino acid sequence to the reference product; any heterogeneity associated with manufacturing conditions that is relevant for safety or efficacy should fall within the range demonstrated for the reference product. This means that the response to the xenogeneic protein sequence is likely to be the dominant factor influencing the human ADA response, not the variables associated with manufacturing process. Unlike the situation for homologous proteins such as epoetins, negative clinical sequalae associated with cross-reactivity of antibodies targeting mAbs have not been reported.
Hypersensitivity reactions (HSR’s) to monoclonal antibodies have been linked to ADAs. Most commonly these are treatment emergent IgG or IgM antibodies. Also Non-ADA related immunologic responses have also been observed for mAbs, e.g. crosslinking Fc receptors, which may lead to injection site reactions. Immediate type reactions occur for nearly all authorized monoclonal antibodies, but these have mostly not been associated with the presence of ADA [19]. A notable exception is cetuximab, where pre-existing anti-galactose-α-1, 3-galactose IgE antibodies have been linked to the occurrence of HSR’s. Severe and fatal infusion reactions occur most often in patients receiving cetuximab for the first time [15]. Known hypersensitivity is a contraindication for the use of cetuximab and HSR’s will likely manifest itself at the first infusion of either innovator or biosimilar, therefore, interchanging is not likely to pose a serious risk for cetuximab. On the contrary, the authors are aware that biosimilar versions of cetuximab are being manufactured using host cell substrates such as CHO, which add much lower levels of non-human glycans such as galactose-α-1,3-galactose. These biosimilar versions might then be expected to have a lower risk of inducing allergic reactions, while maintaining equivalent efficacy/potency to the reference version. In this respect, it is relevant to emphasize that both the EU and US regulatory pathways for biosimilars allow the candidate biosimilar to have lower immunogenicity than the reference product, providing that this does not alter therapeutic efficacy. However, when considering interchangeability, safety issues may occur when switching patients from a lower immunogenic biosimilar product to a higher immunogenic reference product. This could lead to the paradoxical situation that, even though a biosimilar product may be considered safer than the reference product on a population level, it may not be considered interchangeable for safety reasons.
How to ensure continued interchangeability throughout a product’s life cycle
EU legal provisions effectively allow the biosimilar and originator products to pursue independent life-cycles following authorization, including changes in manufacturing process, formulation, primary container and even introduction of new therapeutic indications. This creates a theoretical possibility that post-authorization manufacturing changes – for either the biosimilar or the originator versions – could influence critical product quality attributes in opposite directions (drift) leading to increasingly different products [20].
On the other hand, the analysis of pre- versus post-change product quality attributes, including glycosylation profile, is a mandatory part of the change control process. Effectively, the risk then becomes the uncertainty associated with unexplored or incompletely understood factors such as impact of changes in aggregate/sub-visible particle levels, arising from changes in formulation or primary container, on longer-term clinical manifestations of immunogenicity.
The regulatory approval of such changes to the manufacturing process, or to formulation or primary container, of a biosimilar product in the post-authorization setting is subject to the standards for demonstrating comparability between the pre- versus post-change material, as defined in the ICH Q5E guideline, without evidence to demonstrate continuing biosimilarity to the originator product – or to other biosimilar versions of the same product [20]. The challenge for biosimilar products then becomes the question of how to demonstrate that interchangeability is maintained in the longer-term, particularly following changes to either the originator or (multiple) biosimilar product versions, to an extent that provides sufficient re-assurance that these products may be used interchangeably to achieve an equivalent therapeutic benefit–risk in any individual subject. It is important to state that no post-authorization immunogenicity-related issues have yet been detected in the post-authorization phase for any EU-approved biosimilar product.
How to monitor interchangeability post-authorization?
Although uncertainties that remain from the pre-approval weight of evidence – such as the clinical impact of immunogenicity in different populations in the longer-term clinical experience – may be addressed by commitments made in the EU Risk Management Plan, the studies performed in the post-approval setting are usually non-comparative. Most importantly, there is no mandatory legal obligation to perform clinical studies that directly compare the biosimilar versus originator products in the post-approval period. Nevertheless, the Risk Management Plan of Inflectra/Remsima contains a number of post-marketing commitments, including three additional comparative studies versus Remicade in rheumatoid arthritis and Crohn’s disease.
It has been noted that immunogenicity risk is relatively low beyond 12 months of infliximab therapy: 90% of the patients who develop sustained ADA do so within the first 12 months of therapy (although transient ADA can be detected throughout the duration of infliximab therapy). Also for adalimumab, ADA developed mostly before 12 months of treatment [21]. However, it is challenging to interpret the clinical significance of these data: changes to the management of patients with positive ADA detected after this time point should depend on the clinical setting [22]. ADA assays are prone to interference by residual circulating drug and given the differences in these assays, antibodies can only be assessed in a comparative manner and should not be viewed in isolation of clinically relevant events [23]. If a potential change in immunogenicity after 12 months were considered to represent the most important uncertainty, comparative data using standardized bioanalytical methods would seem essential. To monitor the long-term effect of ADA formation, a centralized laboratory would be required that uses standardized assays as well as correlating the ADA results to measures of drug trough concentration, sustainability of response to treatment and incidence and severity of adverse events.
To assess the continuing interchangeability post-authorization, data would need to be obtained in an ongoing manner. However, there are quite some challenges to using observational safety studies to identify meaningful differences in low frequency events such as hypersensitivity reactions and serious infections. It needs to be kept in mind that these products will be launched in the countries in the European periphery that have higher prevalence of serious infections like tuberculosis (TB), which could confound an accurate interpretation of observational data on infectious safety events [24]. Furthermore, periods for elective switching should also be taken into account, as patients who lose therapeutic response, for example, because of ADA development, may be more likely to be switched between treatments. It will be very challenging to collect appropriate data to answer these questions in a definitive and suitably controlled manner. The Risk Management Plan of Inflectra/Remsima includes two patient registries with a total targeted enrolment of 6,200 patients to evaluate the safety and efficacy in both RA and inflammatory bowel disease indications, with a special focus on serious infections including TB [25]. These registries are projected to produce their final results in 2026.
Currently, authorized TNFIs are studied in a multitude of patient registries worldwide and biosimilar manufacturers are encouraged by EU regulators to contribute to existing biologicals registries [26]. Using observational data to study assess post-authorization safety issues is complex and it will require substantial follow-up to be able to draw meaningful conclusions. While various studies from registries have confirmed the increased risk of serious infections in patients receiving TNFIs [27–29], another recent study did not observe additional risks of serious infections versus standard of care, highlighting the challenges of excluding additional risks for individual products using observational data [30].
Traceability
Switching patients between therapeutically alternative medications repeatedly, without adequate documentation of the specific products and batches administered, may hinder tracing the sources of a potential safety issue. This has been linked to the debate about naming requirements for biosimilars. It has been proposed to assign unique INN (International Nonproprietary Name) suffixes for biosimilars to improve the traceability of potential ADRs, although this proposal has been heavily criticized for being scientifically inconsistent and motivated by marketing considerations [31]. European legislation mandates prescription of all biological medicinal products by brand name, as well as recording of batch number of the product used [32]. For biologicals, brand names were recorded for over 95% of the ADRs reported for products for which biosimilar versions are on the market, although batch numbers are poorly recorded [33]. To ensure the safety of interchanging, ensuring traceability is necessary. Although not an apparent issue for the EU, there may be concern for traceability in other territories that allow prescribing at the INN level.
Conclusions
It will be highly challenging to establish interchangeability of biosimilars to an extent that fully satisfies the US legislative criteria that enable substitution at the pharmacy level. While it may be feasible to demonstrate that treating patients with an alternating regimen shows the same clinical efficacy and safety as patients treated continuously with the reference product, such studies will only be able to address comparability on an aggregated (population) level. Most importantly, they may not be very informative for ensuring continuing post-authorization interchangeability and substitutability of multiple versions of the same product that may be subject to independent changes in conditions of manufacture, formulation or primary container. At the time of registration, not all clinical variables will have been tested, such that uncertainties may remain to be addressed via heightened pharmacovigilance and post-authorization studies. For some products, demonstration of interchangeability/substitutability will be compromised by the uncertain relationship between longer-term antibody formation and clinical outcomes, allied to bioanalytical bias and drug interference. Post-authorization patient registries with enhanced monitoring for immunogenicity, drug levels, sustainability of efficacy and incidence and severity of adverse events would be required to follow up longer-term interchangeability. However, registries have limitations in their ability to provide the necessary evidence to establish long-term benefit/risk and will require a substantial number of patients to be followed up to identify or exclude differences in attributed risk of a biosimilar over its reference product.
While monitoring the safety of biosimilars post-authorization is a standard feature of the EU and US regulatory provisions, suitable processes must be applied to ensure traceability. Even though no incremental safety risks have been identified to date in the post-authorization setting for EU-approved biosimilars, it may be prudent to involve the prescribing physician in a decision to switch patients between therapeutic alternatives. Increasing uptake of biosimilars into medical practice would then take account of the disease activity and therapeutic response of individual patients, as well as financial incentives to make treatment choices. The rigorous regulatory approach for marketing authorization that assures therapeutic equivalence at the outset would be subject to the moderating influence of the prescribing physician to adapt treatment according to individual needs. Arguably, creating a regulatory basis for interchangeability criteria would impose an unreasonable burden to demonstrate continuing biosimilarity in comparative clinical studies – thereby defeating the cost savings achieved to the public health system.
Many of the longer-term concerns about continued interchangeability create considerable practical challenges and many post-authorization activities add little definitive information about the safety and efficacy of biosimilars, which is in the end the prime concern of prescribers and patients. Successfully completing a biosimilarity exercise will establish that two products are therapeutically equivalent; there has been no reason to assume that the act of interchanging such products has any effect on patient outcome. Discriminating between interchangeable and non-interchangeable will disqualify those products for which it is hard to demonstrate interchangeability, or that are currently marketed without the status of interchangeability. Furthermore, the challenge will be to satisfy the US condition that an interchangeable product that can be ‘expected to produce the same clinical result as the reference product in any given patient’. Upon successfully completing a biosimilarity exercise it may be expected that the product produces the same result in all patients. This does not mean that a product will produce the same clinical result as the reference product in every patient. For all these reasons, it should be questioned whether the ‘higher’ standard required for designation of interchangeability/substitutability adds to the benefit of patients.
Competing interests: No funding was received for the preparation of this manuscript. Part of the research of Dr Hans C Ebbers is performed within the context of ‘Escher Projects’ that resides under the umbrella of Dutch public–private partnership Top Institute Pharma (www.ti-pharma.com) and receives funding from the European Federation of Pharmaceutical Industries and Associations (EFPIA) and the Association of the European Self-Medication Industry (AESGP).
Provenance and peer review: Commissioned; externally peer reviewed.
Authors
Hans C Ebbers1,2, PhD
Paul Chamberlain3, BSc
1Utrecht University, Faculty of Science, Utrecht Institute for Pharmaceutical Sciences, Department of Pharmaceutics
2Utrecht University, Faculty of Science, Utrecht Institute for Pharmaceutical Sciences, Department of Clinical Pharmacology and Pharmacoepidemiology
3NDA Advisory Board, NDA Advisory Services Ltd, Prime House, Challenge Court, Barnett Wood Lane, Leatherhead, Surrey KT22 7DE, UK
References
1. Evens RP, Kaitin KI. The biotechnology innovation machine: a source of intelligent biopharmaceuticals for the pharma industry- mapping biotechnology’s success. Clin Pharmacol Ther. 2014 Jan 21. doi: 10.1038/clpt.2014.14
2. Alvogen, Hospira launch Remicade biosimilar in Central, Eastern Europe. Genetic Engineering & Biotechnology News. 2014 Feb 13 [cited 2014 Mar 13]. Available from: http://www.genengnews.com/gen-news-highlights/alvogen-hospira-launch-remicade-biosimilar-in-central-eastern-europe/81249502
3. GaBI Online—Generics and Biosimilars Initiative. Alvogen launches infliximab biosimilar in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2014 Mar 31]. Available from: http://www.gabionline.net/Biosimilars/News/Alvogen-launches-infliximab-biosimilar-in-Europe
4. Ebbers HC, Crow SA, Vulto AG, Schellekens H. Interchangeability, immunogenicity and biosimilars. Nat Biotechnol. 2012 Dec;30(12):1186-90.
5. Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Giezen TJ, et al. Biosimilars: what clinicians should know. Blood. 2012 Dec 20;120(26):5111-7.
6. Shrank WH, Choudhry NK, Agnew-Blais J, Federman AD, Liberman JN, Liu J, et al. State generic substitution laws can lower drug outlays under Medicaid. Health Aff (Millwood). 2010 Jul;29(7):1383-90.
7. U.S. Senate. Biologics Price Competition and Innovation Act of 2009, Public Law 111-148—Mar. 23, 2010 124 Stat. 703;351.
8. Endrenyi L, Chang C, Chow SC, Tothfalusi L. On the interchangeability of biologic drug products. Stat Med.2013 Feb 10;32(3):434-41.
9. U.S. Department of Health and Human Services Food and Drug Administration. Guidance for Industry. Scientific considerations in demonstrating biosimilarity to a reference product. Draft guidance. February 2012 [homepage on the Internet]. 2012 Feb 8 [cited 2014 Mar 31]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
10. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Draft. 03 June 2013 [homepage on the Internet]. 2013 Jun 19 [cited 2014 Mar 31]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/06/WC500144124.pdf
11. Colombel JF, Sandborn WJ, Rutgeerts P, Enns R, Hanauer SB, Panaccione R, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology. 2007 Jan;132(1):52-65.
12. European Medicines Agency. European Public Assessment Report. Retacrit (epoetin zeta) [homepage on the Internet]. [cited 2014 Mar 31]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000872/human_med_001031.jsp&mid=WC0b01ac058001d124
13. Navarro-Millan I, Sattui SE, Curtis JR. Systematic review of tumor necrosis factor inhibitor discontinuation studies in rheumatoid arthritis. Clin Ther. 2013 Nov;35(11):1850-61.e1.
14. Louis E, Mary JY, Vernier-Massouille G, Grimaud JC, Bouhnik Y, Laharie D, et al. Maintenance of remission among patients with Crohn’s disease on antimetabolite therapy after infliximab therapy is stopped. Gastroenterology. 2012 1;142(1):63-70.e5.
15. Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther. 2012 Nov;12(11):1473-85.
16. Flodmark CE, Lilja K, Woehling H, Jarvholm K. Switching from originator to biosimilar human growth hormone using dialogue teamwork: single-center experience from Sweden. Biol Ther. 2013;3:35-43.
17. van Schouwenburg PA, Rispens T, Wolbink GJ. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol. 2013 Mar;9(3):164-72.
18. Scheinberg MA, Kay J. The advent of biosimilar therapies in rheumatology—“O brave new world”. Nat Rev Rheumatol. 2012 Jun 5;8(7):430-6.
19. Baldo BA. Adverse events to monoclonal antibodies used for cancer therapy: focus on hypersensitivity responses. Oncoimmunology. 2013 Oct 1;2(10):e26333.
20. Schneider CK, Vleminckx C, Gravanis I, Ehmann F, Trouvin JH, Weise M, et al. Setting the stage for biosimilar monoclonal antibodies. Nat Biotechnol. 2012 Dec;30(12):1179-85.
21. Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011 Apr 13;305(14):1460-8.
22. Ungar B, Chowers Y, Yavzori M, Picard O, Fudim E, Har-Noy O, et al. The temporal evolution of antidrug antibodies in patients with inflammatory bowel disease treated with infliximab. Gut. 2013 Sep 16. doi: 10.1136/gutjnl-2013-305259
23. Chamberlain P. Assessing immunogenicity of biosimilar therapeutic monoclonal antibodies: regulatory and bioanalytical considerations. Bioanalysis. 2013 Mar;5(5):561-74.
24. World Health Organization. Global tuberculosis report 2013 [homepage on the Internet]. 2014 Oct 24 [cited 2014 Mar 31]. Available from: http://apps.who.int/iris/bitstream/10665/91355/1/9789241564656_eng.pdf?ua=1
25. European Medicines Agency. European Public Assessment Report. Inflectra [homepage on the Internet]. 2013 Oct 4 [cited 2014 Mar 31]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002778/human_med_001677.jsp&mid=WC0b01ac058001d124
26. Zink A, Askling J, Dixon WG, Klareskog L, Silman AJ, Symmons DP. European biologicals registers: methodology, selected results and perspectives. Ann Rheum Dis. 2009 Aug;68(8):1240-6.
27. Dixon WG, Hyrich KL, Watson KD, Lunt M, Galloway J, Ustianowski A, et al. Drug-specific risk of tuberculosis in patients with rheumatoid arthritis treated with anti-TNF therapy: results from the British Society for Rheumatology Biologics Register (BSRBR). Ann Rheum Dis. 2010 Mar;69(3):522-8.
28. Curtis JR, Xie F, Chen L, Baddley JW, Beukelman T, Saag KG, et al. The comparative risk of serious infections among rheumatoid arthritis patients starting or switching biological agents. Ann Rheum Dis. 2011 Aug;70(8):1401-6.
29. van Dartel SA, Fransen J, Kievit W, Flendrie M, den Broeder AA, Visser H, et al. Difference in the risk of serious infections in patients with rheumatoid arthritis treated with adalimumab, infliximab and etanercept: results from the Dutch Rheumatoid Arthritis Monitoring (DREAM) registry. Ann Rheum Dis. 2013 Jun;72(6):895-900.
30. Grijalva CG, Chen L, Delzell E, Baddley JW, Beukelman T, Winthrop KL, et al. Initiation of tumor necrosis factor-α antagonists and the risk of hospitalization for infection in patients with autoimmune diseases. JAMA. 2011 Dec 7;306(21):2331-9.
31. The INN crowd. (editorial) Nat Biotechnol. 2013 Dec;31(12):1055.
32. European Commission. Directive 2010/84/EU. Official Journal L 348, 31/12/2010, p. 74-99.
33. Vermeer NS, Straus SM, Mantel-Teeuwisse AK, Domergue F, Egberts TC, Leufkens HG, et al. Traceability of biopharmaceuticals in spontaneous reporting systems: a cross-sectional study in the FDA Adverse Event Reporting System (FAERS) and EudraVigilance databases. Drug Saf. 2013 Aug;36(8):617-25.
34. European Commission. What you need to know about biosimilar medicinal products. Process on corporate responsibility in the field of pharmaceuticals access to medicines in Europe. A consensus information document [homepage on the Internet]. 2013 Apr 21 [cited 2014 Mar 31]. Available from: http://ec.europa.eu/enterprise/sectors/healthcare/files/docs/biosimilars_report_en.pdf
35. Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008 Feb;117(2):244-79.
36. Baert F, Noman M, Vermeire S, Van Assche G, D’Haens G, Carbonez A, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med. 2003 Feb 13;348(7):601-8.
|
Author for correspondence: Hans C Ebbers, PhD, Utrecht University, Faculty of Science, Utrecht Institute for Pharmaceutical Sciences, Department of Pharmaceutics, PO Box 80 082, 3508 TB Utrecht, The Netherlands |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2014 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



Hello there!
Of some reason the figures, tables and the references related to this paper are not uploading as expected.
Please advise how can I get the full articles (to include the abovementioned items).
Kind regards,
CP
Dear Dr Cornel Pater,
We very much appreciate your kind feedback. The figures, tables and references are available on the website article.
Thank you for your interest in GaBI. Please enjoy the quality information and content published under GaBI (GaBI Online and GaBI Journal).
GaBI Journal Editorial Office