Pathway to affordable, quality-assured sources of pegylated interferon alpha for treating hepatitis C
Published on 2013/09/30
Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(4):194-203.
Author byline as per print journal: Barbara Milan, Sara Gaspani
|
Introduction: The current pipeline of promising oral hepatitis C drugs could lead to a revolution in treatment for this disease in both developed and developing countries. At present, the recommended treatment is pegylated interferon alpha (in combination with ribavirin). However, the limited availability and high cost of this medicine is a major barrier to expanding access to treatment in developing countries. |
Submitted: 12 August 2013; Revised: 21 October 2013; Accepted: 1 November 2013; Published online first: 14 November 2013
Introduction
Hepatitis C virus (HCV) is a growing public health concern. HCV infection affects an estimated 150-180 million people worldwide and kills about 350,000 annually [1, 2]. The disease burden is greatest in developing countries, with the highest reported prevalence in Egypt (22%), Pakistan (4.8%) and China (3.2%) [3]. In 2010, the World Health Assembly (WHA) issued a resolution (the WHA63.18 on viral hepatitis) which, among other actions, urged Member States to provide access to pharmaceutical products and to use national legislative mechanisms that overcome patent barriers for hepatitis C medicines [4].
The current recommended treatment for HCV is pegylated interferon alpha-2a and 2b, a biological product administered by injection once a week, in combination with oral ribavirin [5, 6]. While there has been some skepticism that such a long, injection-requiring treatment is feasible in resource-poor settings, a recent systematic review has documented that treatment with pegylated interferon in low- and middle-income countries is feasible and that outcomes are comparable to those achieved in high income countries [7]. However, the availability and cost of the innovator pegylated interferon alphas are major barriers to access in developing countries, as we will document in this paper.
Since pegylated interferon alpha is, for now, the only effective HCV treatment, in December 2012 Médecins Sans Frontières (MSF) submitted an application for inclusion of this medicine on the WHO Essential Medicines List – a step intended to highlight unmet needs in combatting this epidemic and to trigger policy changes that expand patients’ access to HCV treatment [8]. WHO has included pegylated interferon alpha in the WHO Model List of Essential Medicines released in July 2013 [9], but other steps are needed to secure access to the current recommended treatment – steps that could draw on lessons learned from treatment scale-up in other epidemics, such as HIV [10].
One lesson is that easier-to-use medicines are desperately needed, and these now appear to be on the horizon: over the next few years a promising pipeline of new drugs for hepatitis C should lead to a fully oral treatment [11], which could revolutionize HCV treatment in both developed and developing countries. Another is that, in the interim, expanding access to the current treatment for patients living with the disease is an imperative. A crucial advance towards this goal is that generic versions of pegylated interferon alpha are being developed and/or marketed in several low- and middle-income countries including Cuba, Egypt, India and Iran. However, since competition is blocked in high-income countries by the existence and enforcement of patents [12], none of these products has yet been evaluated by, or gained approval from, a stringent regulatory authority.
This paper summarizes some key findings and approaches in the search for feasible, accessible alternatives to the current out-of-reach price of innovator pegylated interferon alpha in low- and middle-income countries. First, we present our analysis of the landscape of biosimilars regulatory guidance and discuss how the current international guidelines impact access to quality-assured sources of pegylated interferon and potentially to other essential biological drugs. Next, we provide an overview of the market of pegylated interferon alpha products, which could be evaluated for safety and efficacy and then potentially used to expand treatment. We also present price information and analysis on the effectiveness of competition in decreasing prices for innovator medicines and making treatment affordable for patients and their governments. While a comprehensive package of HCV care will also require lowering barriers surrounding diagnostics, laboratory tests and equipment, these issues are beyond the scope of this paper.
Methods
We conducted a review of the biosimilars official guidance documents that have been published by WHO, by stringent regulatory authorities like EMA, FDA, and by drug regulatory authorities in selected middle-income countries. This review also involved retrieving published articles and white papers (from both academic and industry biosimilars experts) on the regulatory pathways and challenges associated with developing and marketing biosimilars. We have limited inclusion of articles and reviewed those which particularly targeted aspects impacting on access to quality-assured biosimilars. In particular, we focused on the need of comparability exercise at the clinical level. This paper is not meant to be an exhaustive review.
In parallel, we compiled information on biosimilars and alternative pegylated interferon alpha products marketed in several developing countries reported in Table 1. The identified pharmaceutical companies authorized the publication of data, which is not included in their product label. In 2012, we sourced price information from official wholesalers/distributors of Roche’s pegylated interferon alpha-2a and Merck’s pegylated interferon alpha-2b in a few selected countries, see Table 2, to provide a snapshot of private sector prices. National currencies were converted into US$ using the exchange rate on the date of data collection. Additional price trend data on the negotiated prices for the two innovator products over time (2006–2011) were obtained directly from the Egyptian Hepatitis C Committee in 2011, see Figure 1, to demonstrate how competition can lead to price decreases for innovator products and impact positively on access to HCV treatment overtime.



Results
Biotechnological drugs and biosimilars: the complexity
Biotechnological drugs are large complex molecules and a precise three-dimensional folded structure. They are manufactured by living organisms or cells engineered in highly sophisticated ways, with approximately 90% of all biological products produced from three sources: E. coli, yeast or Chinese Hamster ovary cells [13]. The size and complexity of biological drugs make it almost impossible to manufacture identical replicas, unlike the situation with small molecule drugs, e.g. HIV antiretroviral drugs. Consequently, the scientific community does not define these new products as ‘generic’ versions of innovator ones [14].
However, there is unfortunately no uniform nomenclature; instead, a variety of terms are used inconsistently, leading to confusion and misperceptions that have hindered acceptance of biosimilars among prescribing physicians and patients in high-income countries. In addition to ‘biosimilar’, other terms include ‘similar biotherapeutic product’, ‘follow-on biologic’, ‘similar biological medicinal product’, ‘subsequent entry biologic’, ‘biogeneric’, ‘me-too biologic’, and ‘non-innovator biologic’ [16]. According to the European Medicines Agency (EMA), ‘biosimilar’ is a copy version of an already-approved innovator (reference) product with demonstrated similarity of physicochemical characteristics, efficacy and safety, as determined by a comprehensive comparability study [17]. The same concept of biosimilars is adopted by other entities, although under different terminology – ‘similar biotherapeutic products’ by WHO, ‘follow-on biologics’ in the US (recently switched to ‘biosimilars’) and Japan, and ‘subsequent entry biologics’ in Canada [18]. Biological products that have been structurally and/or functionally altered to achieve an improved or different clinical performance are often referred to as ‘second-generation biologicals’ or even ‘bio-betters’. From a regulatory perspective, the claim of ‘better’ must be substantiated (usually by presenting a full registration dossier) with data showing a clinically relevant advantage over a previous-generation product.
Investments needed to develop biosimilars
Developing biosimilar medicinal products requires considerable investment of time and money. The process generally takes about eight years, far longer than the two years typical for developing small molecule generics, see Figure 2.
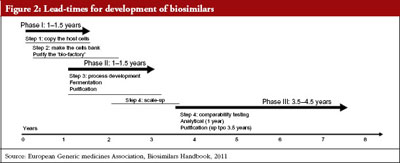
It is also extremely expensive, requiring a pre-launch investment in the range of US$10–US$140 million for a bacterially-produced biosimilar product such as pegylated interferon alpha, compared to US$1–US$5 million for a small molecule generic drug [15], see Figure 3.

As with other types of medicines, biosimilars can be marketed following the expiry of patents on the innovator product. But that is often precisely the time point when second-generation biological drugs (or bio-betters) are also introduced into the market, and may be preferred due to improved product characteristics such as better stability profiles, less frequent treatment schedules, and/or increased safety. This is exactly what occurred when biosimilar products of non-pegylated interferon alpha were ready to enter the market in Europe, pegylated interferons were launched precisely at the same time and quickly became the first choice for treatment, taking over the market space of biosimilar non-pegylated interferons.
With the requirement for huge financial pre-launch investments usually followed by lucrative business in high-income countries, the biosimilar market has been characterized by partnering between large multinational pharmaceutical companies and other technology giants such as Fuji Film and Samsung [19]. Nevertheless, in low- and middle-income countries, such as Cuba and Iran [20], other business models exist whereby smaller biological drugs producers rely on much lower-scale investment and government-financed technology transfer programmes to meet mainly national treatment demands.
Overview of regulatory guidance on biosimilars
The assessment principles used to evaluate biosimilars are more complex than those used for small molecule generics. Due to the complexity of biological drugs, the classical bioequivalence approach used for small molecule pharmaceuticals is not deemed suitable to establish therapeutic equivalence between the biosimilar and the reference (innovator) drug product [21].
EMA: The European Medicines Agency was the first regulatory agency to provide a framework for evaluating biosimilars. Since 2005, it has issued overarching guidelines for biosimilars [22] (undergoing revision [23]), for quality issues relevant to the development of biosimilars [24] (undergoing revision [25]), and for non-clinical and clinical issues [26], (undergoing revision [27]) and immunogenicity [28]. In subsequent years EMA released several product class-specific guidelines as well as draft guidelines or concept papers for product classes, such as human insulin, heparin, somatropin, erythropoietin and monoclonal antibodies [29].
EMA’s comprehensive biosimilar regulatory pathway relies on the principle of comparability exercise at the preclinical and clinical levels, a controversial approach that has triggered a considerable debate. Since 2005, 14 biosimilars of three biological products (somatropin, erythropoietin, and filgrastim) have been approved in the EU [30], several of which were approved with only partial or no direct preclinical and clinical comparison to the corresponding innovator products [31]. Biosimilar producers consider these comparative clinical trials duplicative and responsible for driving up development costs drastically [32]; likewise, some representatives of the scientific community consider preclinical and clinical comparability studies to be ‘surplus’ requirements and have called for them to be dropped [31, 33]. This has a particular relevance considering that additional 75 other products are in development in the European Union [34].
Although EMA reacted strongly to these calls and reiterated the need for comparability exercise at the clinical level [35], the most recent guidance (the 2013 EMA ‘Guideline on non-clinical and clinical development of similar biological medicinal products containing low-molecular-weight-heparins) shows some flexibility on this point [36]. For example, the biosimilars draft guideline [23] appears to challenge the necessity of a full comparability exercise and that the guideline allows some exceptions to this requirement. The guideline states that ‘in specific circumstances, e.g. for structurally more simple biological medicinal products, a comparative clinical efficacy study may not be necessary if similarity of physicochemical characteristics and biological activity/potency of the biosimilar and the reference product can be convincingly shown and similar efficacy and safety can clearly be deduced from these data and comparative pharmacokinetics (PK)
data’ [23].
To facilitate these alternative pathways, i.e. without repetition of clinical trials, EMA created the option of comparing the biosimilar in certain clinical studies and in vivo non-clinical studies (where needed) to an innovator product that was authorized not by EMA but by a regulatory authority with scientific and regulatory standards similar to those of the EMA, i.e. from countries that are members of the International Conference of Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use [23, 37]. Although simple, this change could have profound consequences in terms of easing and shortening the biosimilars approval process. Indeed, trials conducted outside Europe and using other reference products may now be used for EMA filings (unlike in the past, when EMA required repetition of these trials in European patients, using an EMA-approved reference product), thereby leading to significant savings for biosimilar manufacturers [38].
US FDA: In the US, although the Food and Drug Administration (US FDA) pioneered the development and implementation of an abbreviated approval pathway for small molecule generics through the 1984 Hatch-Waxman Act, an abbreviated pathway for licensure of biosimilar products was formally created only in March 2010 [39] under Section 351 of the Public Health Service Act (42 U.S.C. §262) [40].
In February 2012, the US FDA issued three draft documents to guide industry in developing biosimilar products [41–43]. These draft guidelines allow for waivers of preclinical and clinical studies: ‘As a scientific matter, comparative safety and effectiveness data will be necessary to support a demonstration of biosimilarity if there are residual uncertainties about the biosimilarity of the two products based on structural and functional characterization, animal testing, human PK and pharmacodynamics (PD) data, and clinical immunogenicity assessment. A sponsor may provide a scientific justification if it believes that some or all of these comparisons on clinical safety and effectiveness are not necessary’ [41]. However, as underscored by public comments to these documents, the drafts are unclear about which requirements are indispensable for the US FDA and which ones might be discussed on a case-by-case basis [44, 45].
The lack of finalized guidelines discourages biosimilars producers from submitting applications for registration to the US FDA, which has never received a filing for a biosimilars application [46]. US economists estimated that the regulatory approval process will take at least two years, in which case the first biosimilar approved under the new guidance would not enter the US market for at least two years after adoption of US FDA biosimilars guidelines [47].
WHO: In 2009, the World Health Organization published Guidelines on Evaluation of Similar Biotherapeutic Products, defined as a ‘living document’ that should be developed further as scientific knowledge and experience accumulates [48] and that is modelled on the previously published EMA guidelines. Both sets of guidelines advocate a stepwise approach in which licensing a biosimilar depends on demonstrating similarity in terms of quality, non-clinical and clinical parameters, to a reference product (an innovator biological product with the same dosage form and route of administration), that has already been licensed on the basis of a full registration dossier. Under WHO guidelines, the comparability exercise between the biosimilar and the reference product is requested for the quality, non-clinical and clinical parts. If relevant differences between the biosimilar and the reference product are detected at any step, the reasons for these differences must be explored and justified; if this is not possible, a full licensing (stand alone) application should be considered. In a reversal of the usual sequence of events, WHO guidelines on biosimilars actually preceded the WHO draft guidelines on non-clinical and clinical evaluation of the quality, safety and efficacy of biological medicinal products prepared by recombinant DNA technology [49].
Adoption and adaptation of biosimilars/similar biotherapeutic products regulatory guidance in other countries
EMA and WHO guidelines have a strong influence on the biosimilars regulatory framework around the world. Some stringent regulatory authorities, such as that in Australia, adopted the European guidelines [50]. Others, including Canada in 2010 [51], and Japan in 2009, issued new guidelines based on the current EMA guidelines. Since the WHO guideline was released in 2009, 11 National Drug Regulatory Agencies (NDRAs) have published their own guidelines for registering biosimilars/similar biotherapeutic products [13], these NDRAs are from Brazil, Iran, Jordan, Malaysia, Mexico, Singapore, Republic of South Korea, Saudi Arabia, South Africa, Turkey [52], and India [53]. There are differences among criteria adopted by different countries [54]. Indeed some NDRAs such as in Brazil, Mexico, South Korea, India and Iran adapted the general WHO guidance to their specific country context, generally with lower barriers in terms of clinical trial requirements [55].
For example, the Brazilian 2010 guidelines foresee two pathways for similar biotherapeutic products approval [56]: the comparative pathway, based on the principles outlined by WHO; and an individual development pathway with a reduced dossier comprising a full package of data on quality but reduced non-clinical and clinical studies [57].
The 2009 South Korea biosimilar guidelines are also flexible: ‘if the comparability can be demonstrated by confirmatory PK/PD data, an efficacy study may be omitted’ [58]. Similarly, the Indian ‘similar biologic’ guidelines published in August 2012 allow waiving of the comparative clinical trial under certain conditions [53]. Likewise, the Iranian NDRA does not require comprehensive new preclinical or clinical data to prove similarity between locally produced biological drugs and innovator products [59].
A number of countries allow yet another approach to defining the reference product, given that the innovator product may have not been (and may never be) registered in a country. For example, Iran allows evaluation of a domestically produced biological drug against a US FDA or EMA approved biosimilar product [52]. In India, if the reference biological drug is not licensed domestically, it is permitted to use a reference product that is licensed in another country with a well-established regulatory framework and that has been widely marketed for at least four years (or less in cases of a national healthcare emergency) [53, 60].
Innovator and alternative pegylated interferon alpha products
Pegylated interferon alpha-2b (PEG-Intron, Merck) was registered by EMA in May 2000 [61] and by US FDA in January 2001 [62]. Pegylated interferon alpha-2a (Pegasys, Hoffmann-La Roche) was registered by EMA in June 2002 [63] and by US FDA in October 2002 [64].
PEG-Intron, Merck, pegylated interferon alpha-2b is a covalent conjugate of recombinant alpha-2b interferon (approximate molecular weight [MW] 19,271 daltons) with monomethoxy polyethylene glycol (PEG, MW = 12,000 daltons). The average molecular weight of the PEG-Intron molecule is approximately 31,000 daltons. Interferon alpha-2b is produced by recombinant DNA techniques from the fermentation of an E. coli strain that harbours a genetically engineered plasmid with an interferon gene from human leukocytes [65].
Pegasys, Hoffmann-La Roche, pegylated interferon alpha-2a, is a covalent conjugate of recombinant alpha-2a interferon (approximate MW 20,000 daltons) with a single branched bis-monomethoxy polyethylene glycol (PEG) chain (approximate MW 40,000 daltons). The PEG moiety is linked at a single site to the interferon alpha moiety via a stable amide bond to lysine. Pegylated interferon alpha-2a has an approximate MW of 60,000 daltons. Interferon alpha-2a is produced using an engineered E. coli strain containing a cloned human leukocyte interferon gene [66].
Pegylated interferon alpha products are being developed and marketed in several low- and middle-income countries, including Cuba, Egypt, India and Iran. Table 1 lists the products we identified during our search for alternatives to the innovator products. A few products seem to have been developed as copies of Pegasys, or PEG-Intron in terms of molecular structure and in certain instances also using the same excipients in the same proportions. Other products represent a different size and form of PEG branches and different attachment sites between the PEG moiety and the molecule of interferon alpha-2a or 2b.
Whether or not the innovator products are patented, has in turn, influenced the ways that producers in low- and middle-income countries have approached development of their products. While the total absence of patents on pharmaceuticals in Iran has made it possible to develop and scale up production of a replica of Roche’s pegylated interferon alpha-2a, in India this molecule was under patent. As a result, the Indian ‘similar biologic’ market has focused on replicas of Merck’s pegylated interferon alpha-2b, due to the absence of patents on the molecule. However, a patent on the innovator’s formulation of pegylated interferon alpha-2b drove development in India towards an alternative formulation, in this case with different excipients, to avoid patent infringement.
Some manufacturers have opted to use a host cell other than E. coli. According to the WHO 2009 guidance [48], ‘the host cell type for manufacture of the [biosimilar] should only be changed if the manufacturer can demonstrate convincingly that the structure of the molecule is not affected or that the clinical profile of the product will not change’. Similarly, a change of excipients is permitted under the EMA guidelines provided that this change makes the formulation suitable for stability, compatibility, integrity, and activity and that its potential impact on safety and efficacy is appropriately justified [25]. For example, the Indian company Zydus Cadila has opted to develop pegylated interferon alpha-2b using a different host cell system (Pichia pastoris yeast instead of E. coli) to simplify the process, and to change excipients as a way of circumventing patent protection. Another Indian company, Virchow, has developed a pegylated interferon version that is reportedly similar in profile to PEG-Intron, which has led to litigation with Merck over the use of certain excipients in the formulation [12]; Virchow has also developed alternative formulations with different excipients.
Pricing trends of the originators products and why competition is important
In 2012, the range of product prices from the two innovator companies at wholesaler/distributor level range from US$200 to US$375 per vial or pre-filled syringe in Eastern Europe, Central Asia and India, see Table 2. Prices increase further at the level of private retail pharmacies and therefore out of reach to most patients in the private sector. This further confirms previous survey findings [11], with the two innovator companies selling the originator medicines at similar prices in the same countries.
The countries surveyed here have no governmental HCV treatment programme, so treatment is available only within the private sector. Average patient income in these countries does not allow spending the sums needed to buy these medicines, even excluding diagnostic and laboratory monitoring costs.
In 2013, Roche started marketing pegylated interferon alpha-2a in India under the new brand name Exxura and has decreased its price by more than 50% in one year, to US$125/vial. Similarly Merck’s product is now available for roughly the same price. The presence of alternative products, such as those from Virchow and Zydus Cadilla has also led private sector prices to drop over the last year, but still does not make them an affordable treatment option.
Egypt, one of the countries hit hardest by the hepatitis C epidemic, stands as an exception. Since its establishment in 2006, the National Hepatitis C Committee has managed to progressively reduce the price of pegylated interferon alpha and ribavirin combination therapy, see Figure 1. The Egyptian Ministry of Health has succeeded in negotiating progressive price reductions with both Roche and Merck, from US$79 for pegylated interferon alpha alone in 2006 to US$41 for one vial of pegylated interferon plus weekly supply of ribavirin in 2011 – bringing the 48-week treatment cost to about US$2,000 per patient. This decrease was triggered by the introduction in the Egyptian market of an alternative pegylated interferon alpha, which has reduced the country’s dependence on the multinational companies Roche and Merck and created a more competitive environment [67, 68].
Discussion
The two innovator companies of pegylated interferon alpha have been marketing their products at very similar prices in both developed and developing countries. The collected data shown here indicate the very high prices charged by the companies in low- and middle-income countries, and which make these medicines out of reach for the vast majority of patients. The data also show that when alternative drugs are available, for example, in Egypt and India, the innovator companies have lowered the price of their product significantly. It is urgent that Merck and Roche adopt a preferential pricing policy so that HCV treatment can be expanded and scaled up in the public and private sectors in low- and middle-income countries.
Given the crucial role in decreasing prices of biosimilars marketed in developing countries, plus the long lead-times for developing new ones, it is critical that the existing drugs (the additional identified sources of pegylated interferon alpha other than the innovator drugs which have been evaluated by stringent regulatory authorities) are evaluated for safety and efficacy by an independent body such as WHO.
The majority of developing countries have little or no regulatory capacity in the area of biological drugs, nor do non-governmental organizations (NGOs) have the capacity to assess biological drugs (as opposed to small molecule generics) using international standards. Countries and NGOs therefore need to rely on international evaluation frameworks to access safe, quality-assured pegylated interferon, for example, via a WHO pre-qualification system for biological drugs/biosimilars that enables member states to access to safe and effective pegylated interferon alpha and other essential biological medicines. The WHO pre-qualification system for HIV medicines had a huge impact in securing quality-assured generic antiretroviral medicines, to allow for competition in the market space, decrease prices tremendously and therefore to allow scale up of treatment in developing countries [10].
Today, WHO and the regulatory authorities are confronted by the key challenge of comparability exercise at the clinical level, and of how manufacturers can meet this requirement despite a much lower level of investment than in the European biosimilars industry. The compelling but unmet needs for targeted biological drugs in developing markets have fostered local regulatory processes with reduced requirements for comparative clinical trials [53, 56–59, 69]. Biosimilars regulatory guidance should be reviewed in light not only of the scientific and regulatory experience gained over time, but also of the needs and interests of national health systems and pharmaceutical markets in low-resource countries [70]. Stringent regulatory authorities such as EMA have already begun to waive requirements for comparability exercise at clinical level under appropriate circumstances. This approach is supported by academic experts who claim that non-comparative clinical trials are sufficient for regulatory purposes, and who call for pragmatic approaches focused primarily on the patients clinical outcomes and on scientific principles, using the state-of-the-art tools [31, 33, 71].
Conclusion
The current WHO guidelines on similar biotherapeutic products should be reassessed based on accumulated science and experience, and in ways that foster the development and marketing of quality-assured, safe and effective biological drugs. In parallel, a WHO-supported programme for evaluating pegylated interferon alpha products is needed to ensure competition, and consequently affordable prices and sustainable medicines supplies. Without such an evaluation scheme, the addition of pegylated interferon alpha to the essential medicines list will have only limited impact, even in countries where political will and financial resources are present. Preferential pricing policies by the innovator companies are also needed, but they will not be able to satisfy all demands for initiating and scaling up HCV treatment across different countries and organizations. Competition with quality-assured sources of pegylated interferon alpha products is the key to expand access to HCV treatment with pegylated interferons either in combination with ribavirin or with the oral promising antiviral drugs coming onto the market. Therefore, there is a need to support regulatory work in WHO in the biosimilars field both in guidance definition and in product evaluation for essential biological medicines.
Acknowledgements
We gratefully acknowledge Mr Karen Gharagyozyan for the survey of innovator pegylated interferon alpha prices in countries of Eastern Europe and Central Asia, Ms Patricia Kahn for editing of the manuscript, Ms Elena Rosso and Mr Rizwan Ahmed for providing information on the Indian prices of pegylated interferon alpha, Ms Suzette Kox (EGA) for authorization to reproduce Figure 2, Mr Mishra Neelkanth, Mr Aggarwal Anubhan, Mr Walton Jo (Credit Suisse) for authorization to reproduce Figure 3, the pharmaceutical companies that authorized the publication of data beyond the label of their pegylated interferon alpha product and finally the academic and industry experts who provided valuable input into this project.
Disclosure of financial and competing interests: The authors are employed by the Médecins Sans Frontières’ Access Campaign and they declare no conflict of interest. Neither the authors Ms Barbara Milani and Ms Sara Gaspani, nor their organization have received any type of funding, incentives or in-kind payments and donations from pharmaceutical companies or have any vested interest in the use of any particular product belonging to the category of innovator or of off-patent biopharmaceuticals presented in this paper.
Médecins Sans Frontières (MSF) is an international, independent, medical humanitarian organization that delivers emergency aid to people affected by armed conflict, epidemics, natural disasters, and exclusion from healthcare. In 1999, in the wake of MSF being awarded the Nobel Peace Prize, MSF launched the Access Campaign. Its purpose has been to push for access to, and the development of life-saving and life-prolonging medicines, diagnostic tests and vaccines for patients in MSF programmes and beyond.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors are responsible for English language editing of this manuscript.
Co-author
Sara Gaspani, Project Pharmacist, Médecins Sans Frontières, Access Campaign
References
1. World Health Organization. Prevention and control of viral hepatitis infection: framework for global action. Geneva: 2012 [homepage on the Internet]. [cited 2013 Oct 21]. Available from: http://www.who.int/csr/disease/hepatitis/GHP_Framework_En.pdf
2. Hadigan C, Kottilil S. Hepatitis C virus infection and coinfection with human immunodeficiency virus: challenges and advancements in management. JAMA. 2011 Jul 20;306(3):294-301.
3. Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005 Sep;5(9):558-67.
4. World Health Organization. World Health Assembly Resolution 63.18 on viral hepatitis. Geneva: 2010. [homepage on the Internet]. [cited 2013 Oct 21]. Available from: http://apps.who.int/gb/ebwha/pdf_files/WHA63/A63_R18-en.pdf
5. EASL. Clinical practice guidelines: management of hepatitis C virus infection [homepage on the Internet]. J Hepatol. 2011;55(2):245-64.
6. National Institute for Health and Clinical Excellence. Peginterferon alpha and ribavirin for the treatment of chronic hepatitis C: part review of NICE technology appraisal guidance 75 and 106. London, September 2010 [homepage on the Internet]. 2011 Jun [cited 2013 Oct 21]. Available from: http://www.nice.org.uk/nicemedia/live/13180/50856/50856.pdf
7. Ford N, Kirby C, Singh K, et al. Chronic hepatitis C treatment outcomes in low- and middle-income countries: a systematic review and meta-analysis. Bull World Health Organ. 2012 Jul 1;90(7):540-50.
8. Médecins Sans Frontières Access Campaign. Submission for the inclusion of peginterferon alpha-2a and -2b in the WHO Essential Medicines List [homepage on the internet]. 2013 [cited 2013 Jun 13]. Available from: http://www.msfaccess.org/content/msf-submission-inclusion-peginterferon-alpha-2a-and-2b-who-essential-medicines-list
9. World Health Organization. WHO model list of essential medicines: 18th list, April 2013 [homepage on the Internet]. 2013 Jul [cited 2013 Oct 21]. Available from: http://www.who.int/medicines/publications/essentialmedicines/18th_EML_Final_web_8Jul13.pdf
10. Ford N, Singh K, Cooke GS, et al. Expanding access to treatment for hepatitis C in resource-limited settings: lessons from HIV/AIDS. Clin Infect Dis. 2012 May;54(10):1465-72.
11. Médecins Sans Frontières Access Campaign. Diagnosis and treatment of hepatitis C: a technical landscape [homepage on the Internet]. 2013 Apr [cited 2013 Oct 21]. Available from: http://www.msfaccess.org/sites/default/files/MSF_assets/HepC/Docs/HepC_Report_DxTxTech_ENG_2013_FINAL.pdf
12. Initiative for Medicines Acces and Knowledge. Patent Landscape report for pegylated interferon alfa 2a and 2b [homepage on the Internet]. 2013 [cited 2013 Oct 21]. Available from: http://www.i-mak.org/storage/I-MAK%20 Patent%20 Landscape%20for%20Pegylated%20Interferon%20 Alfa%202 A%20and%202B.pdf
13. Dranitsaris G, Amir E, Dorward, K. Biosimilars of biological drug therapies: regulatory, clinical and commercial considerations. Drugs. 2011 Aug 20;71(12):1527-36.
14. Schellekens H. Biosimilar therapeutics-what we need to consider? NDT Plus. 2009;2[Suppl 1]: i27-i36.
15. Mishra N, Aggarwal A, Walton J, et al. Biosimilar 101. Credit Suisse Report. 2009 August.
16. Wadhwa M, Thorpe R. Terminology for biosimilars–a confusing minefield. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(3-4):132-4. doi:10.5639/gabij.2012.0103-4.023
17. Weise M, Bielsky MC, De Smet K, et al. Biosimilars-why terminology matter. Nat Biotechnol. 2011;29(8):690-3.
18. Hincal F. An introduction to safety issues in biosimilars/follow-on biopharmaceuticals. J Med CBR Def. 2009;7:1-17.
19. Carrol J. Everyone wants in, but with no ground rules, the biosimilars business remains elusive.Biotechnol Healthc. 2011 Fall; 8(3):12-3.
20. International Centre for Genetic Engineering and Biotechnology. Activity report 2010/2011 [homepage on the Internet]. [cited 2013 Oct 21]. Available from: www.icgeb.org/research-groups.html?file=tl_files/hq/…11.pdf
21. Schellekens H. Assessing the bioequivalence of biosimilars. The Retacrit case. Drug Discov Today. 2009 May;14(9-10):495-9.
22. European Medicines Agency. Guideline on similar biological medicinal products. CHMP/437/04 [homepage on the Internet]. 2005 Oct [cited 2013 Oct 21]. Available from: www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf
23. European Medicines Agency. Draft revised of guideline on similar biological medicinal products. CHMP/437/04 Rev 1 [homepage on the Internet]. 2013 May [cited 2013 Oct 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/05/WC500142978.pdf
24. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: quality issues. EMEA/CHMP/BWP/49348/2005 [homepage on the Internet]. 2006 Jun [cited 2013 Oct 21]. Available from: www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003953.pdf
25. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1) [homepage on the Internet]. 2012 May [cited 2013 Oct 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf
26. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: non clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005 [homepage on the Internet]. 2006 Jun [cited 2013 Oct 21]. Available from: www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf
27. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: non clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005 Rev. 1 [homepage on the Internet]. 2013 Jun [cited 2013 Oct 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/06/WC500144124.pdf
28. European Medicines Agency. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. EMEA/CHMP/BMWP/14327/2006 [homepage on the Internet]. 2007 Feb [cited 2013 Oct 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003947.pdf
29. European Medicines Agency. Scientific guidelines on biosimilar medicines [homepage on the Internet]. 2013 [cited 2013 Oct 21]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000408.jsp&mid = WC0b01ac058002958c
30. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe. [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Oct 21]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
31. Schellekens H, Moors E. Clinical comparability and European biosimilar regulations. Nat Biotechnol. 2010 Jan;28(1):28-31.
32. McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs. 2011 Mar-Apr;3(2):209-17.
33. Schellekens H, Moor EHM. Reply: in support of the European Union biosimilar framework. Nat Biotechnol. 2012;30(8):748-9.
34. Datamonitor Healthcare. EU biosimilars: driving global sales growth reference code: HC00237-015. 2012 Aug [cited 2013 Oct 21]. Available from: https://service.datamonitorhealthcare.com/company/biosimilars/hot-topics/article90150.ece
35. Schneider CK, Borg JJ, Ehmann F, et al. In support of the European Union biosimilar framework. Nat Biotechnol. 2012; 30(8):745-8.
36. European Medicines Agency. Guideline on non- clinical and clinical development of similar biological medicinal products containing low-molecular-weight-heparins [homepage on the Internet]. 2013 Jan [cited 2013 Oct 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/01/WC500138309.pdf
37. European Medicines Agency. European Medicines Agency to accept biosimilar reference medicines sourced outside European Economic Area [homepage on the Internet]. 2012 Sep 28 [cited 2013 Oct 21]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2012/09/news_detail_001615.jsp&mid=WC0b01ac058004d5c1
38. European Medicines Agency. Commissioner Dalli delivers speech on healthy and active ageing through improved access to affordable generic medicines, June 2012 [homepage on the Internet]. 2012 Jun [cited 2013 Oct 21]. Available from: http://ec.europa.eu/commission_2010-2014/dalli/docs/speech_15062012.pdf
39. U.S. Food and Drug Administration. Biologics Price Competition and Innovation Act of 2009, Pub. L. 111-148, Sect. 7001-7003, 124 Stat. 119 [homepage on the Internet]. 2010 Jun [cited 2013 Oct 21]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/ucm216146.pdf
40. U.S. Food and Drug Administration. Public Health service Act Se. 262. Regulation of biological products [homepage of the Internet]. 2013 [cited 2013 Oct 21]. Available from: http://www.fda.gov/RegulatoryInformation/Legislation/ucm149278.htm
41. U.S. Food and Drug Administration. Draft guidance for industry. Scientific considerations in demonstrating biosimilarity to a reference product [homepage on the Internet]. 2012 Feb [cited 2013 Oct 21]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
42. U.S. Food and Drug Administration. Draft guidance for industry. Quality considerations in demonstrating biosimilarity to a reference protein product [homepage on the Internet]. 2012 Feb [cited 2013 Oct 21]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf
43. U.S. Food and Drug Administration. Draft guidance for industry. Biosimilars: questions and answers regarding implementation of the Biologics Price Competition and Innovation Act of 2009 [homepage on the Internet]. 2012 Feb [cited 2013 Oct 21]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM273001.pdf
44. Biotechnology Industry Organization. Comments on quality considerations in demonstrating biosimilarity to a reference protein product FDA Docket No. FDA–2011-D-0602 [homepage on the internet]. 2012 Apr [cited 2013 Oct 21]. Available from: www.bio.org/sites/default/files/2012-04-16 Biosimilars Quality Considerations – FINAL.pdf
45. Hunton & Williams LLP. ClientAlert. FDA issues three guidances on biosimilar product development [homepage on the Internet]. 2012 Feb [cited 2013 Oct 21]. Available from: http://www.hunton.com/files/News/48b1d25c-2ad9-4f57-a949-0bf2354e3939/Presentation/NewsAttachment/cd585ad5-2b74-416d-bd28-0f6d7d815b63/fda_issues_three_draft_guidances.pdf
46. Rotenstein LS, Ran N, Shivers JP, et al. Opportunities and challenges for biosimilars: what’s on the horizon in the global insulin market? Clin Diabetes. 2012;30(4):138-50.
47. Blackstone EA, Fuhr JP Jr. Innovation and competition: will biosimilars succeed?: The creation of an FDA approval pathway for biosimilars is complex and fraught with hazard. Yes, innovation and market competition are at stake. But so are efficacy and patient safety. Biotechnol Healthc. 2012 Spring;9(1):24-7.
48. World Health Organization. Expert Committee on Biological Standardisation, Geneva, 19 to 23 October 2009. Guidelines on evaluation of Similar Biotherapeutic Products (SBPs) [homepage on the Internet]. 2010 Jun [cited 2012 Oct 21]. Available at: http://www.who.int/biologicals/areas/biological_therapeutics/biotherapeutics_for_web_22april2010.pdf
49. World Health Organization. WHO guidelines on the quality, safety and efficacy of biological medicinal products prepared by recombinant DNA technology non clinical and clinical evaluation [homepage on the Internet]. 2013 Mar [cited 2013 Oct 21]. Available from: http://www.who.int/biologicals/WHO_rDNA_1 st_Public_consultation_12_March_2013.pdf
50. European Medicines Agency. Guideline on similar biological medicinal products. CHMP/437/04. London, 30 October 2005 [homepage on the Internet]. 2006 Jun [cited 2013 Oct 21]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf
51. Health Canada. Guidance for sponsors: information and submission requirements for Subsequent Entry Biologics (SEBs), 2010 [homepage on the Internet]. 2013 [cited 2013 Oct 21]. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/applic-demande/guides/seb-pbu/seb-pbu_2010-eng.php
52. Hadavand N, Valadkhani M, Zarbakhsh A. Current regulatory and scientific considerations for approving biosimilars in Iran. Biologicals. 2011 Sep;39(5):325-7.
53. Central Drugs Standard Control Organization. Guidelines on similar biologics: regulatory requirements for marketing authorisation in India [homepage on the Internet]. 2012 Jun [cited 2013 Oct 21]. Available from: http://cdsco.nic.in/Bio%20Similar%20Guideline.pdf
54. Kang HN. Summary of the diverse situation of similar biotherapeutic products in the selected countries (August 2010). Biologicals. 2011 Sep;39(5):304-7.
55. GaBI Online – Generics and Biosimilars Initiative. Positioning of biosimilars: commodity versus differentiated. [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2013 Oct 21]. Available from: www.gabionline.net/layout/set/print/Reports/Positioning-of-biosimilars-commodity-versus-differentiated
56. Castanheira LG, Barbano DB, Rech N. Current development in regulation of similar biotherapeutic products in Brazil. Biologicals. 2011 Sep;39(5):308-11.
57. da Silva Madeira L, Borschiver S, Pereira Jr N. Prospects and trends in the Brazilian market for biologically sourced products. J Technol Manag Innov. 2012;7(3):44-56.
58. Korea Food and Drug Administration. Guidelines on the evaluation of biosimilar products. 2010. p.34
59. Cheraghali AM, Current status of biopharmaceuticals in Iran’s pharmaceutical market. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(1).26-9. doi:10.5639/gabij.2013.0201.008
60. Ohly C. The New India Guidelines on similar biologics regulatory and market authorization requirements. October 2012 [homepage on the Internet]. [cited 2013 Oct 21]. Available from: http://fr.slideshare.net/brandsynapse/the-newindiabiosimilarguidelines
61. European Medicines Agency. PEG-Intron authorisation details [homepage on the Internet]. 2013 [cited 2013 Oct 21]. Available from: www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000395/human_med_000974.jsp&mid=WC0b01ac058001d124
62. U.S. Food and Drug Administration. PEG-Intron Label and Approval Information. Drugs@FDA. [homepage on the Internet]. [cited 2013 Oct 21]. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm
63. European Medicines Agency. Pegasys authorisation details [homepage on the Internet]. 2013 [cited 2013 Oct 21]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000395/human_med_000974.jsp&mid=WC0b01ac058001d124
64. U.S. Food and Drug Administration. Pegasys Label and Approval Information. Drugs@FDA. [homepage on the Internet]. [cited 2013 Oct 21]. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm
65. U.S. Food and Drug Administration. Label PEG-intron (pegylated interferonalpha-2b) powder for injection [homepage on the internet]. 2001 Aug [cited 2013 Oct 21]. Available from: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/Therapeutic
BiologicApplications/ucm094490.pdf
66. U.S. Food and Drug Administration. BLA 103964/0 PEGASYS, peginterferon alfa-2a (Ro 25-8310 Injectable Solution, 180µg/mL) [homepage on the Internet]. 2003 Jul [cited 2013 Oct 21]. Available from:http://www.fda.gov/downloads/Drugs/Development
ApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm094464.pdf
67. Egyptian Initiative for Personal Right. The Egyptian interferon: a scientific debate and necessary regulations that need to be issued [homepage on the Internet]. 2013 [cited 2013 Oct 21]. Available from: http://eipr.org/en/report/2011/09/21/1258
68. Egyptian Ministry of Health & Population. National Committee for Control and Prevention of Viral Hepatitis. Letter to Médecins Sans Frontières (MSF) Egypt Mission. July 2012.
69. Egyptian Drug Regulatory authority. Draft guidelines on registration of biosimilar products [homepage on the Internet]. 2012 Dec [cited 2013 Oct 21]. Available from: http://www.eda.mohealth.gov.eg/Download/Docs/Final%20biosimilar%20 guideline.pdf
70. Cheraghali AM, Biosimilars; a unique opportunity for Iran national health sector and national pharmaceutical industry, Daru. 2012 Sep 10;20(1):35. doi:10.1186/2008-2231-20-35.
71. Oldfield P, A wide angle view of biosimilars from a bioanalytical perspective. Bioanalysis. 2013 Mar;5(5):533-5. doi:10.4155/bio.12.316.
|
Author for correspondence: Barbara Milani, Pharmaceutical Coordinator, Médecins Sans Frontières, Access Campaign, 78 Rue de Lausanne, PO Box 116, CH-1211 Geneva 21, Switzerland |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2013 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



Where to buy pegylated interferon alpha in Malaysia, sarawak.