Pharmaceutical Data Integrity: issues, challenges and proposed solutions for manufacturers and inspectors
Published on 2020/10/08
Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(4):171-82
Author byline as per print journal: Adjunct Associate Professor Sia Chong Hock1, BSc (Pharm), MSc; Vernon Tay1, BSc (Pharm) (Hons); Vimal Sachdeva2, MSc; Associate Professor Chan Lai Wah1, BSc (Pharm) (Hons), PhD
|
Abstract: |
Introduction
Data Integrity (DI) in the pharmaceutical manufacturing industry is the state where data are Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available (ALCOA+) [1–3], as outlined in Table 1. Data altered such that it no longer fulfils these criteria is considered as falsified, regardless of it being due to human error or generated deliberately [2, 4].
Current legislation, good manufacturing practice (GMP) standards and guidance on data management and governance published by organizations such as the US Food and Drug Administration (FDA) [6–8] and World Health Organization (WHO) [1] aim to guide the industry in ensuring DI is not compromised. These include the ‘Data Integrity and Compliance with cGMP Guidance for Industry’ from FDA [9], ‘GxP Data Integrity Guidance and Definitions’ from the UK Medicines and Healthcare products Regulatory Agency (MHRA) [10], and ‘Guidance on Good Data and Record Management Practices’ from WHO [1], which were published in recent years. Inspectors from various organizations inspect the pharmaceutical manufacturing companies to assure compliance to such legislation, standards and guidance, where appropriate [3, 11, 12]. If violations of regulatory significance are observed, warning letters containing the key violations to be rectified would be sent to the companies [13]. However, with the number of FDA warning letters issued citing DI violations quintupling from 2014 to 2017 [14], and large pharmaceutical companies getting cited for falsifying data in quality control results and other manufacturing processes, the effectiveness of such legislation and guidance to maintain DI remains yet to be seen [15, 16].
With an increasing use of computerized systems in the pharmaceutical industry [17, 18], and current regulation of physical data being more well-defined than regulation of electronic data [19], it is uncertain if the legislation and guidance are still able to maintain DI as more electronic data are generated. Furthermore, the outsourcing of pharmaceutical manufacturing activities to improve productivity and business efficiency continues unabatedly [20]. A lack of synergy and good data management between companies increases the difficulty in standardizing protocols and procedures to assure DI [21], regardless of the legislation and guidance in place [22]. Additionally, protocols which help maintain DI in parent companies may not be adopted by their subsidiary companies [23]. Failure to prevent DI violations could lead to substandard medicinal products being released into the market, thus causing harm and possibly death to patients [24, 25] and, in the case of vaccines and biosimilars, loss of public confidence.
Hence, this paper strives to assess the prevalence and trends of recent DI violations, identify reasons why companies commit DI violations, evaluate the effectiveness of current legislation, guidance and challenges, and finally, explore solutions which can promote DI in the pharmaceutical and biopharmaceutical manufacturing industry. A systematic, scientific and comprehensive literature review, covering the websites of regulatory authorities, scientific journals, pharmaceutical fora and newsletters, national and international legislation, GMP and other good practices and guidance documents relating to DI, was conducted. Challenges and issues relating to DI were identified, and solutions to address them were proposed for the benefit of the manufacturers, inspectors and the global pharmaceutical and biopharmaceutical community in general.
Current trends
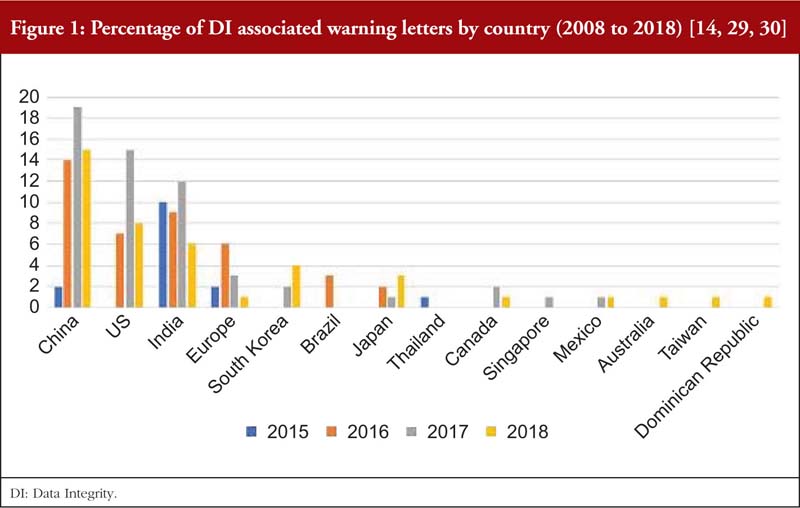
As reported by the Unger Consulting Incorporation [14], the prevalence of FDA warning letters that cited DI violations has been increasing exponentially, see Figure 1. This may be due to pharmaceutical inspectors proactively searching for DI violations [26], inspectors who are now better trained to detect DI issues, more companies taking risks in violating DI for various reasons, or ignorance and carelessness of operators [27]. It is not easy to analyse the root causes of DI violations as the increasing prevalence of DI issues and efforts to manage them appear to be a recent development [28].
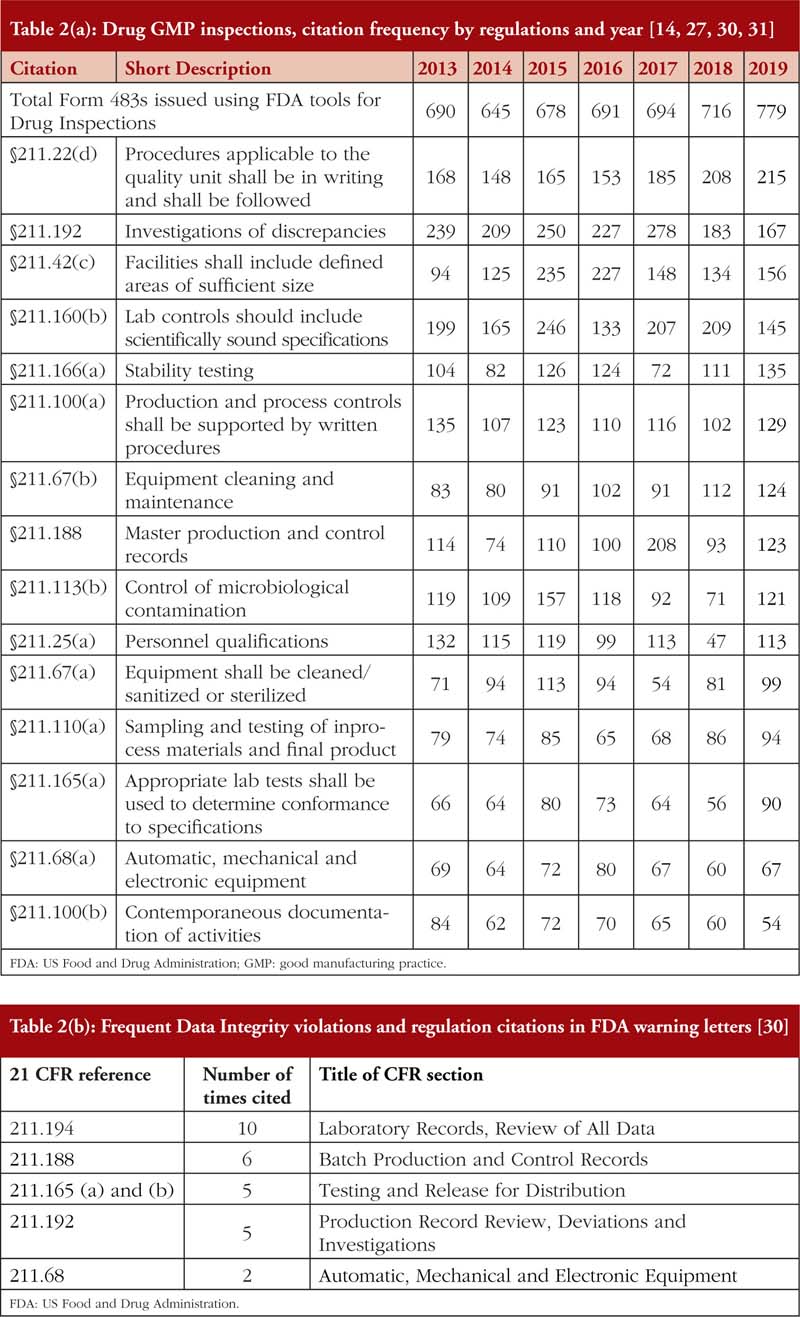
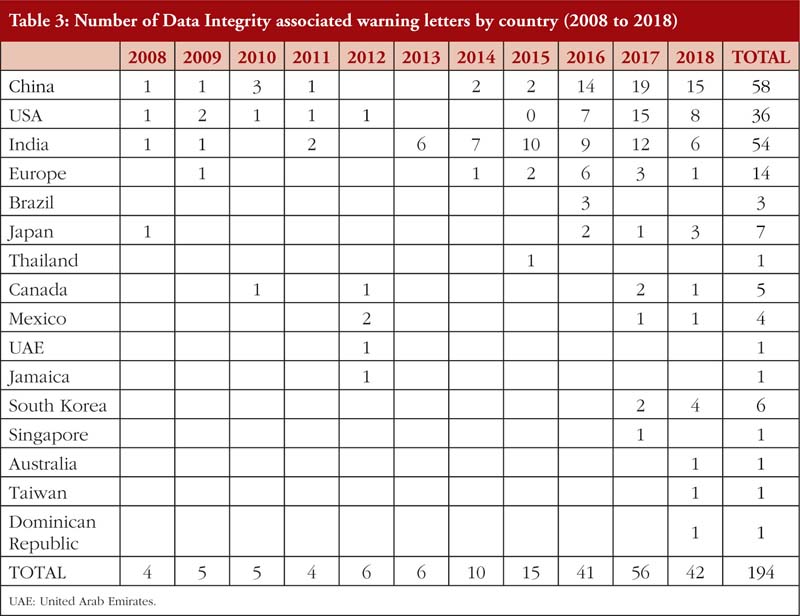
Also, from Tables 2(a) and 2(b), it is noted that most of the DI violations cited pertain to manual, automatic, mechanical and electronic equipment, which includes ‘failure to calibrate and maintain written records’ and ‘failure to exercise appropriate controls over computer or related systems to assure that only authorized personnel institute changes in production and control records, laboratory records or other records’ [31]. The next few most cited DI violations pertain to quality control of the pharmaceutical product. The leading countries being issued DI associated warning letters include China and India [14], see Table 3, where parent pharmaceutical manufacturers in Europe and the US have been known to translocate their manufacturing plants to these countries to reduce production costs [20, 23]. It is also important to emphasize that DI violations are also routinely cited by FDA during inspections of domestic manufacturers as well.
Reasons for Data Integrity violations (inadvertent and intentional)
Pharmaceutical companies are often under pressure to improve their key performance indicators (KPIs), especially during economic downturns. Hence, data are known to be falsified to decrease the rejection of manufactured batches, with some companies deleting non-compliant records [32–36], or even churning out records without legitimately performing relevant tests to expedite regulatory approval [28, 34]. Furthermore, the lack of support from senior management, due to insufficient involvement and resources, can aggravate the situation. Also, some employees may fear retrenchment due to unachieved KPIs [37, 38]. Thus, they may release the product without following internal protocols requiring them to seek approvals from authorized personnel [33], or alter records if given access to the database [35, 39]. Occasionally, and in particular, for systems involving manual transfer of data to the company database using hybrid computerized systems, transcription errors can occur, leading to inaccurate data records [40].
An example of a hybrid approach is where laboratory analysts use computerized instrument systems that create original electronic records and then print a summary of the results. Where hybrid approaches are used, appropriate controls for electronic documents, such as templates, forms and master documents, that may be printed, should be available. However, during on-site inspections of the laboratory systems, it has been discovered that data were being falsified on an industrial scale, using a variety of means, such as copy and paste, manipulation of weights, and unauthorized manual integration of chromatograms. The root cause is often a chromatographic data system (CDS) whose audit trail had been deliberately turned off, and therefore, cannot track who had falsified what data, and when [41].
Assuring and promoting Data Integrity via legislation and guidance documents
Legislation
In this article, regulations from the FDA and the European Union (EU) EudraLex, are discussed and compared. As DI in pharmaceutical manufacturing is strongly associated with GMP, it is important to understand the GMP regulatory framework and its impact on DI. The GMP legislative framework from FDA comprise the 21 CFR 210, 211, 212, 600, and 820, while those from EU comprise Commission Directive 2003/94/EC and its regulatory statute EudraLex Volume 4 [42]. 21 CFR 210 provides a very generic regulation on the safety, identity, strength, quality and purity of pharmaceutical products [7]. EU Commission Directive 2003/94/EC gives a general overview of GMP for the pharmaceutical manufacturing companies [43]. 21 CFR 211 and EudraLex Volume 4 are similar, regulating the required documentation for personnel qualifications and training, equipment protocols, inspections and maintenance, labelling and distribution processes, and even protocols for recalls, and corrective and preventive actions (CAPA) [8, 43], with EudraLex Volume 4 dedicating Chapter 7 to contract requirements for outsourced manufacturing activities [43], whereas such requirements are not explicitly stated in 21 CFR 211. 21 CFR 212 and 600 regulate specifically radiological [44] and biological pharmaceutical products [45], respectively. In general, they require more accurate and attributable information to be kept for a longer time to retrace and recall when issues pertaining to the manufacture of the product arise. 21 CFR 820 dictates requirements to ensure quality is maintained throughout the manufacturing process, specifying the documentations required to validate such processes [46]. Clearly, these legislations cover many aspects where proper documentation and DI should be enforced.
There are also legislation specifically promoting DI in pharmaceutical manufacturing. For example, 21 CFR 11 specifically targets requirements for electronic documentation, stating that these electronic documentations are as significant as paper records, and in certain cases can be used in lieu of them [6]. It regulates computerized systems which have external personnel authorized to access and edit, and systems that are employed within the company. EU Falsified Medicines Directive regulates the labelling and distribution practices of drugs, namely ensuring that packaging cannot be tampered without being noticed, the identity of the contents in the packaging are accurate and attributable, and documentation that assures Internet sales of pharmaceutical products is validated by relevant authorities [47]. The Drug Data Management Standard of China, as translated by the China Working Group of Rx-360, provides regulations to promote DI [48]. It regulates documentation of various processes such as training of personnel, validation of computerized systems and data management, and CAPA when DI violations are found. One section specifically provides examples on how DI would be maintained using ALCOA+ as a guide. In Article 7, it specifically promotes whistleblowing as part of the culture for pharmaceutical manufacturing companies as well [48].
Guidance documents
As legislation tend to be generic to facilitate application to a wide variety of pharmaceutical companies, guidance documents have been published to clarify legislative requirements [49]. In general, guidance documents encourage voluntary compliance and can be adapted to suit the company’s culture and manufacturing processes. Guidance documents published to promote GMP include the WHO Guidance on Good Data and Record Management Practices (WHO Technical Report Series 996, Annex 5) [1], FDA Data Integrity and Compliance with CGMP Guidance for Industry, MHRA GxP Data Integrity Guidance and Definitions, the Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (PIC/S) Guide to Good Manufacturing Practice for Medicinal Products [50], PIC/S Guide to Good Manufacturing Practice for Active Pharmaceutical Ingredients [51], the latter is equivalent to the International Council on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q7 – Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients [52]. The WHO Guidance on Data and Record Management Practices also promotes a company culture of integrity and provides links to relevant legislation and guidance documents [1]. The PIC/S Guide to GMP for Active Pharmaceutical Ingredients has been established for many years already and it addresses the same issues as ICH Q7 [53, 54].
There are also guidance that clarify specific portions of the GMP, including PIC/S Good Practices for Computerized Systems in Regulated GxP Environment [55], FDA Standardization of Data and Documentation Practices for Product Tracing Guidance for Industry [56], FDA Contract Manufacturing Arrangements for Drugs: Quality Agreements – Guidance for Industry [57], and ICH Q9 – Quality Risk Management [58]. These documents provide in-depth guidance to the various aspects of GMP, with due consideration for the respective country’s regulation. However, as mentioned earlier in this paper, there are some guidance that specifically focus on DI. These include the PIC/S Good Practices for Data Management and Integrity in Regulated GMP/GDP Environment [59], FDA Data Integrity and Compliance with cGMP Guidance for Industry [9], and MHRA ‘GxP’ Data Integrity Guidance and Definitions [60], all recently published due to increasing attention on DI [3]. Generally, these guidance documents discuss audit requirements, personnel responsibility in promoting DI, and validation of computerized systems and other GMP processes. The WHO, PIC/S and FDA further provide clarification on CAPA to be taken when DI violations are found [9, 59], with PIC/S providing added clarification on outsourced processes and promotion of quality culture [59]. Some guidance documents also help companies to understand the legal requirements. For example, the Orange Guide [61] compiles relevant legislation and guidance notes for manufacturers planning to enter the UK pharmaceutical market, increasing the ease for manufacturers to understand the regulations by providing relevant guidance and binding legislation.
Overall, the legislation and guidance documents appear to be comprehensive in assuring and promoting DI. However, they are unable to prevent DI violations alone. Most DI issues only surface during on-site audits [18] or from whistleblowing [15], and by then, non-compliant pharmaceutical products would have already been distributed, with potentially substandard products having been consumed by patients. Hence, legislation and guidance must be supplemented with other approaches to promote and assure DI at a higher level.
Proposed solutions to better promote and assure Data Integrity
Culture of integrity
A study was conducted by the Parenteral Drug Association (PDA) to assess the effectiveness of its published DI guidance document. Although more than 90% found this guidance helpful in promoting DI, some remarked that a culture of integrity is required to truly attain DI [62], Incentives, including recognition for companies if no DI issues have been found for a consecutive number of years, could be introduced to encourage companies to follow the guidance.
As mentioned in some legislation and guidance documents, a culture of integrity is required in a company to make regulations work [62]. Setting a culture of integrity is important so that management would treat DI seriously [3, 63], and employees would then feel obligated to do the same [64]. According to a study by Yang, Sun and Eppler, for any strategy to be implemented successfully, the formulation needs to be of a certain standard, and inter- and intra-department relationships should be cordial [62]. Middle management is noted to be the main drivers for implementation [62], and close collaboration with the top management increases its effectiveness [62]. However, if management ignores the DI issues, implementation would be hindered [65]. Open and supportive communication between employees and management aid in effective strategy implementation [62, 66]. Providing internal whistleblowing opportunities to flag any DI issues will further promote a company’s culture of integrity [67, 68]. This method is adopted by FDA, where under the Dodd-Frank Wall Street and False Claims Act, monetary rewards are used to promote whistleblowing behaviour [67].
Having a culture of integrity within the company will reduce DI issues, and ultimately bring about a positive perception of the company’s pharmaceutical products [63]. However, for a large company, it is difficult to start a culture of integrity if this culture was absent in the first place, as the implementation of such a culture requires some time before the effects are fully felt [65]. Furthermore, old habits may cause top management to resist adopting such a culture unless specific incentives are provided [65]. Therefore, some regulations should be in place to start this culture of integrity within the company [64].
Database management systems
Another proposed solution to promote DI is by having good and effective database management. A database management system (DBMS) stores data [69] and presents them in an understandable format when accessed [112]. With data becoming larger in volume and variety in the pharmaceutical manufacturing industry [70], user-friendly and efficient DBMS are in high demand [71]. With validated DBMS, manufacturers and regulators would be better able to focus on other DI-related issues.
This paper also evaluates some of these DBMS, and their effectiveness in promoting DI below. Specifically, the advantages and complications of three major categories of DBMS are compared, see Figure 2.
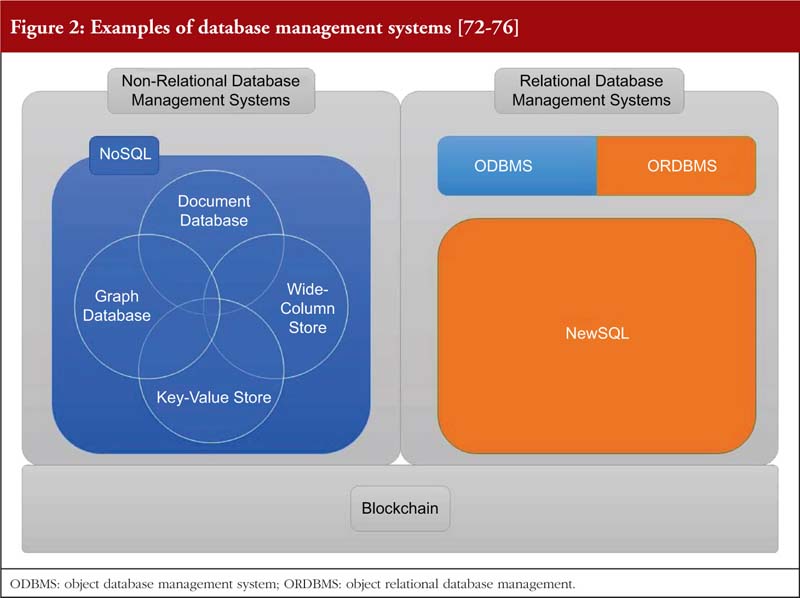
Relational and non-relational database management system
Relational database management systems store data in either a two-dimensional table or a three-dimensional ‘object’ [72, 77]. Non-relational database management system on the other hand does not have a specified structure of storing data. Further elaboration is provided in Tables 4, 5 and 6.
With a wide variety of DBMSchoices currently in the market, adopting one that keeps data ALCOA+ throughout its lifespan would minimize the cost required to maintain it manually [95].
Blockchain technology
Blockchain is hypothesized as the next pharmaceutical manufacturing DBMS innovation [96, 97]. It is a decentralized record of digital events, with validation by the participants occurring before it is recorded [98], making manipulation of previously verified transactions including data entry or movement very hard, and cannot be deleted [99]. Blockchain has three main ways to ensure data security. Firstly, it has a hash function, which identifies blocks, and calculation of hashes involves the previous block’s hash [100]. Secondly, it has a peer-to-peer network to verify before it is added to the current blockchain as a legitimate block [97], removing the need for an authorized person for approval of transaction [98]. Once a block is added, it is added to all the copies of the verified blockchain across the entire network [101], hence remaining in the system indefinitely. Thirdly, as only pre-approved participants can participate in adding new blocks, the identity of the node adding the block would be documented [102], which ensures data attributability.
Furthermore, by using blockchain-utilizing smart contracts, DI can be enforced [103], using blockchain technology to ensure all components of the contract are met before transactions such as approvals occur [103, 104]. This can also be employed for auditing as well, where, if certain values deviate from the acceptable range, they would be flagged up for inspection [100]. Companies such as BlockVerify [103] and One Network Enterprises [96] have started to employ blockchain to maintain DI in the pharmaceutical market. Blockchain has also been applied in promoting DI in the distribution of pharmaceutical products through modium.io AG [105], making use of an array of sensors to ensure erroneous data would not be entered into the system in the first place [103]. In the future, blockchain could even be used to supplement guidance documents [106, 107].
However, handling large volumes of information and simultaneous transactions is slow with current blockchain technology [108], and with more data being generated in pharmaceutical manufacturing companies, this translates to lower efficiency of maintaining DI for large data stores. Furthermore, the difficulty in comprehending and using the code gives the developer the power to maintain DI [109], rendering both authorities and companies incapable of maintaining DBMS DI themselves. Additionally, having a private blockchain requires data encryption [109] to protect data from unauthorized access [107, 110].
In general, having a good DBMS promotes DI as it streamlines audits. Guidance in the form of questions are available to help companies find the best DBMS options available for them [75]. Furthermore, it is common to use multiple databases for different functions [90]. However, relying on DBMS alone will not prevent all DI issues. Firstly, DBMS is unable to ensure data entered was ALCOA-plus, and audits are required to ascertain that [111]. Secondly, unvalidated or outdated systems require upgrading, and migration of data while updating may cause errors to be carried forward unknowingly, especially for large volume of data [95, 112], leading to an inaccurate database. Therefore, DBMS alone cannot prevent all DI issues. Continually upgrading DBMS by periodically reviewing documentation and procedures which influence the quality of pharmaceutical products manufactured against established standards would aid in promoting DI in the future as well [113–116]. However, audits take resources to perform, and smaller companies might not be able to perform frequent and comprehensive self-audits. Nonetheless, having such audits would ensure that the DBMS employed by the company are current and efficient, ultimately promoting DI [95, 114].
Education and training
It is important for employees to undergo DI education and training for them to understand the importance of maintaining DI and the consequences if not maintained [117, 118]. More detailed sessions should be conducted for employees with access to modify processes, systems and records, further explaining their responsibilities [114, 118]. Training sessions could also standardize the procedures, terminology and concepts within the company, reducing DI violations due to miscommunication [114]. Currently, DI courses from external service providers such as ECA Academy [119] and Reading Scientific Services Ltd (RSSL) [120] exist.
However, training can be costly, especially to smaller companies. To mitigate part of the cost, a representative could be trained, before training their fellow colleagues, causing a multiplying effect. Furthermore, manufacturers may form associations together, getting group discounts from DI training providers [120]. To ensure knowledge retention of the training provided, constant refreshers are needed, be it refresher courses or incentivize maintaining DI with company culture, otherwise, such knowledge might be forgotten if infrequently used [121].
Robust quality agreements
With an increase in outsourcing of pharmaceutical manufacturing processes, quality agreements, which are written contracts between companies to ensure responsibilities and expectations for both parties are agreed upon [122], must be rigorously prepared, mutually agreed and signed. However, this process can be time-consuming as reaching a consensus can be challenging. Some guides, such as one from Rx-360 [123], help expedite this process. By having a concise understanding of expectations, the contract giver would hence be able to assure that practices which promote DI would be performed by the contract acceptor, while the contract acceptor understands what is expected of them [124].
Collaboration between countries
Each country has its own set of legislation. Although the legislation of different countries generally overlaps [125, 126], individual countries may not accept specific documents that originate from another country, exacerbating DI issues. Hence, collaborative use of legislation and mutual recognition schemes can help to promote DI [127, 128], with the added benefit of efficient international transactions as DI criteria would have been fulfilled prior to application for regulatory or other transaction approvals.
Effective and efficient audits and audit trail review
Audits are defined as validation checks conducted on manufacturing protocols and systems that assure quality in the processes, products, and computerized systems at the manufacturing site [129]. This includes both internal audits self-conducted by the manufacturer in accordance with Chapter 9 of the PIC/S GMP Guide [130], and external audits conducted by regulatory auditors including FDA and MHRA. As audits are limited in duration, meaningful and efficient audits should be conducted [124]. In general, processes or data that do not affect product safety and compliance, including data on accounting and finances [131], need not be audited. The audit can be streamlined by tagging relevant items to allow the auditor to quickly sieve them out for scrutiny [132, 133]. Audit trails may be divided into two different types: Data Audit Trail (DAT), which covers the raw data recorded, and System Audit Trails (SAT), which covers the systems in place to maintain DI during documentation.
When auditing DAT, critical quality attributes (CQA) and audit trail elements must be defined before conducting the audit. CQAs are the characteristics or properties that can harm patients if not properly controlled [117]. These attributes are to be defined by the company, referring to current legislation and guidance such as ICH Q8(R2) Part 2 [134]. Audit trail elements are the items which affect CQAs and are to be audited [132]. Other items need not be audited as frequently nor meticulously [131]. When auditing SAT, assuming the current system is validated, auditing for possible indicators of DI breaches can substitute an audit of the raw SAT data [131]. These indicators include multiple login attempts and read and write errors [135]. With the recent focus of audits being more SAT-oriented, coupled with more robust systems that can detect errors in DAT [131], falsification of data points prior to documentation may not be detected. As such, both DAT and SAT must be audited in tandem to achieve a more comprehensive audit outcome.
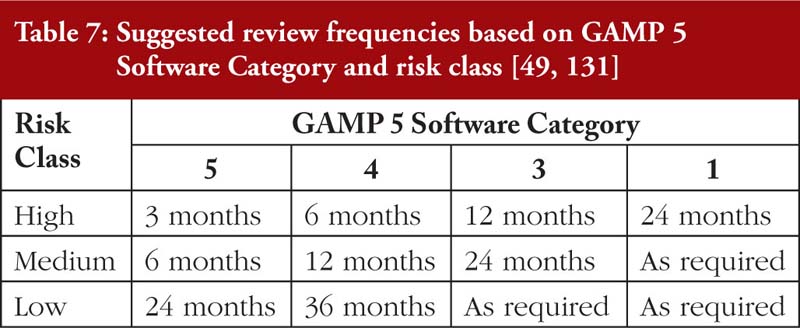
Finally, it must be emphasized that a reliance on periodic audits from the regulator is grossly inadequate to address DI issues. On the other hand, overly frequent internal audits may inefficiently use the company’s manpower. For SAT, an approach based on the risks and implications of DI breaches and the Good Automated Manufacturing Practices (GAMP 5) Software Category [131], as tabulated in Table 7, is recommended.
Computerized systems validation
Pharmaceutical and biopharmaceutical manufacturers should validate their computerized systems such that they are fit for their intended purpose, and to ensure that adequate controls are in place to facilitate tracking and detection of deleted or altered data. The use of hybrid (paper and computerized) systems should be discouraged. However, where legacy systems are awaiting replacement, mitigating controls should be put in place. In such cases, original records generated during the course of GxP activities must be complete and must be maintained throughout the records retention period in a manner that allows the full reconstruction of the GxP activities. Replacement of hybrid systems should be a priority [41].
Conclusion
With increasingly complex pharmaceutical manufacturing processes, maintaining DI might become more challenging, and relying merely on legislation and guidance to maintain DI might be insufficient. Some possible solutions to tackle this challenge include having a company culture of integrity, having a good DBMS, education and training, forming effective quality agreements, collaborations between countries, and performing efficient audits. Together with existing legislation and guidance, these measures can help manage DI issues in the pharmaceutical manufacturing industry, improve the standard of pharmaceutical manufacturing worldwide, and ultimately, produce safe and quality medicinal products for patients internationally.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Adjunct Associate Professor Sia Chong Hock1, BSc (Pharm), MSc
Vernon Tay1, BSc (Pharm) (Hons)
Vimal Sachdeva2, MSc
Associate Professor Chan Lai Wah1, BSc (Pharm) (Hons), PhD
1Department of Pharmacy, National University of Singapore, 18 Science Drive 4, Singapore 117543
2Technical Officer (Senior Inspector), World Health Organization, Prequalification Team, Regulation of Medicines and Other Heath Technologies (RHT), Essential Medicines and Health Products (EMP), Health Systems and Innovation, 20 Avenue Appia, CH-1211 Geneva 27, Switzerland

References
1. World Health Organization. Technical Report Series, Annex 5. Guidance on good data and record management [homepage on the Internet]. [cited 2020 Sep 15]. Available from: practices http://www.who.int/medicines/publications/pharmprep/WHO_TRS_996_annex05.pdf
2. Snee RD. LLC Data integrity – how to detect lack of integrity. IVT Validation Week; 25-27 October 2015; Philadelphia, PA. Available from: https://www.researchgate.net/profile/Ron_Snee/publication/282879414_Data_Int egrity_Validation/links/5684106b08aebccc4e0fdadb.pdf
3. Boogaard P, Haag T, Reid C, Rutherford M, Wakeham C. Data Integrity – here, large, and not going away, here, large, and not going away. Pharmaceutical Eng. 2016;36:41-67.
4. Perez JR. Field notes: maintaining Data Integrity. Quality Progress. 2017; 50(3):14-5.
5. Lydon B. Data Integrity. International Society of Automation. 2017.
6. U.S. 21 Code of Federal Regulations § 11, 1997.
7. U.S. 21 Code of Federal Regulations § 210, 1978.
8. U.S. 21 Code of Federal Regulations § 211, 1978.
9. U.S. Food and Drug Administration. Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Center for Veterinary Medicine (CVM), Data Integrity and compliance with CGMP guidance for industry. April 2016 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.fda.gov/media/97005/download
10. Medicines and Healthcare products Regulatory Agency. GxP Data Integrity Guidance and Definitions. 2018 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/687246/MHRA_GxP_data_integrity_guide_March_edited_Final.pdf
11. Kumar S, Tanwar D, Arora N. The role of regulatory GMP audit in pharmaceutical companies. Int J Res Dev Pharm Life Sci. 2013;2:493-8.
12. Milmo S. Harmonizing GMP inspections, Pharm Tech. 2018;42(5).
13. The FDA Group. The emergency guide to FDA warning letters and FDA 483 – How to handle inspectional observations and warning letters from the FDA. 2017 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: http://www.thefdagroup.com/hubfs/Downloads/The_Emergency_Guide_to_FDA_ Warning_Letters_and_FDA_483.pdf
14. Unger BW. An analysis of 2017 FDA warning letters on Data Integrity. Pharmaceutical Online. 2018.
15. Ranbaxy whistleblower reveals how he exposed massive pharmaceutical fraud, CBS This Morning. 2013.
16. Dooley B. Viral post inflames public anger in China vaccine scandal, Sino Daily. 24 July 2018.
17. Markarian J. Modernizing pharma manufacturing. Pharm Tech. 2018;42:20-5.
18. Tabersky D, Woelfle M, Ruess J-A, Brem S, Brombacher S. Recent regulatory trends in pharmaceutical manufacturing and their impact on the industry. CHIMIA Int J Chem. 2018;72(3):146-50.
19. Unger BW. Data Integrity and data management for GXP regulated firms. Unger Consulting. 2016.
20. Langer ES. Outsourcing trends in biopharmaceutical manufacturing. Pharmaceutical Outsourcing. 24 September 2017.
21. Privett N, Gonsalvez D. The top ten global health supply chain issues: perspectives from the field. Oper Res Health Care. 2014;3(4):226-30.
22. Osani JE. Supplier quality management: monitoring the quality and compliance of api suppliers, defining generic pharma as the customer [thesis]. 2010.
23. Rauschnabel J. 2018 Pharma Industry Outlook – the importance of quality and safety, especially in times of change. Contract Pharma. 2018.
24. Burns M. Management of narrow therapeutic index drugs. J Throm Thrombolysis. 1999;7(2):137-43.
25. Eldawud R, Stueckle T, Manivannan S, Elbaz H, Chen M, Rojanasakul Y, et al. Real-time analysis of the effects of toxic, therapeutic and sub-therapeutic concentrations of digitoxin on lung cancer cells. Biosens Bioelectron. 2014;59:192-9.
26. Ruth A. Why pharmaceutical Data Integrity is more important than ever. Pharmaceutical Guidelines. 2017.
27. Mullin R. The drug industry’s data integrity problem. Chem Eng News. 2018;96(2):15-7.
28. Pharma Quality Europe. EU and US joint inspections: data as the cornerstone for the future. FDAnews’ 8th FDA Inspections Summit. 2018 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.fdanews.com/ext/resources/files/Conference/FIS13Presentations/DIncerti–EU-and-US-Joint-Inspections-Data-as-The-Cornerstone-for-The-Future.pdf
29. U.S. Food and Drug Administration. Warning letters. 2018 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.fda.gov/drugs/warning-letters-and-notice-violation-letters-pharmaceutical-companies/warning-letters-2018
30. Bioprocess Online: FDA FY2019 Drug inspection observations and trends. 6 December 2019.
31. Unger BW. Warning letters 2016 – Data governance and Data Integrity.
32. U.S. Food and Drug Administration. Warning letters. Wockhardt Limited 7/18/13. https://www.ipqpubs.com/wp-content/uploads/2014/04/2013-_-Wockhardt-Limited-7_18_13.pdf
33. U.S. Food and Drug Administration. Warning letter. Marck Biosciences Ltd. July 08, 2014 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm409898.htm
34. U.S. Food and Drug Administration. Warning letters. Mahendra Chemicals July 13, 2015 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm455345.htm
35. U.S. Food and Drug Administration, Warning letters. Sri Krishna Pharmaceuticals Ltd. – Unit II. April 1, 2016 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.fda.gov/iceci/enforcementactions/warningletters/2016/ucm495535.htm
36. U.S. Food and Drug Administration. Warning letters. Zhejiang Ludao Technology Co., Ltd. 23 February 2018 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm598585.htm
37. Vandekerckhove W. Freedom of expression as the “broken promise” of whistleblower protection, Revue Des Droits De l’Homme. 2016;10. doi:10.4000/revdh.2680.
38. Kierans L, Lewis D. Using statutory guidance and codes of practice to build on whistleblowing legislation: the Irish experience. Revue Des Droits De l’Homme. 2016;10. doi:10.4000/revdh.2716.
39. U.S. Food and Drug Administration, Warning letters. Jilin Shulan Synthetic Pharmaceutical Co. Ltd. May 5, 2018 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm608713.htm
40. DiPalma D, Vannest J. Integrated systems aid Data Integrity, Pharmaceutical Technology. 2018;42(5):46-7.
41. McDowall RD. Data Integrity focus, Part III: what is the problem with hybrid systems? LCGC North America. 2019;37(3):180-4.
42. European Commission. Commission Directive 2003/63/EC. Official Journal of the European Union. 2003 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol- 1/dir_2003_63/dir_2003_63_en.pdf
43. European Commission. The rules governing medicinal products in the European Union. Office for Official Publications of the European Communities, Luxembourg, 1998. 2003 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://ec.europa.eu/health/documents/eudralex/vol-4_en
44. U.S. 21 Code of Federal Regulations. § 212, 2009.
45. U.S. 21 Code of Federal Regulations. § 600, 2014.
46. U.S. 21 Code of Federal Regulations. § 820, 1996.
47. Directive 2011/62/EU of The European Parliament and of The Council of 8 June 2011 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2011_62/dir_2011_62_en.pdf
48. SFDA Drug Data Management Standard. 2016 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://rx-360.org/wp-content/uploads/2018/08/SFDA-Drug-Data-Management-Standard-English-Version.pdf
49. Embrey MA. Chapter 6: Pharmaceutical legislation and regulation, in: MDS-3: Managing Access to Medicines and Health Technologies. Kumarian Press; 2013.
50. Guide to good manufacturing practice for medicinal products, Pharmaceutical Inspection Convention, Pharmaceutical Inspection Co-operation Scheme, Geneva, Switzerland, 2009.
51. Guide to good manufacturing practice for active pharmaceutical ingredients, Pharmaceutical Inspection Convention, Pharmaceutical Inspection Co-operation Scheme, Geneva, Switzerland, 2009.
52. The International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients, Q7, 2000. [cited 2020 Sep 15]. Available from: https://database.ich.org/sites/default/files/Q7%20Guideline.pdf
53. API cGMP facility questionnaire & audit checklist, n.d. [cited 2020 Sep 15]. Available from: https://www.coursehero.com/file/35614050/Q7A-API-GMP-Questionnairedoc/
54. Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co- operation Scheme. Revised PIC/S audit checklist based. 2014 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.picscheme.org/layout/document.php?id=675
55. Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co- operation Scheme. Good practices for computerized systems in regulated GXP environment. 2007 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.picscheme.org/layout/document.php?id=155
56. U.S. Food and Drug Administration. Standardization of data and documentation practices for product tracing guidance for industry. 2018 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM598734.pdf
57. U.S. Food and Drug Administration. Contract manufacturing arrangements for drugs: quality agreements. Guidance for Industry. 2016 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm353925.pdf
58. The International Council on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, Q9 Quality risk management. 2005 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: [https://database.ich.org/sites/default/files/Q9%20Guideline.pdf
59. Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co- operation Scheme. Good practices for data management and integrity in regulated GMP/GDP environments. 2016 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://picscheme.org/layout/document.php?id=714
60. Medicines and Healthcare products Regulatory Agency. ‘GXP’ Data Integrity Guidance and Definitions. 2018 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attac hment_data/file/687246/MHRA_GxP_data_integrity_guide_March_edited_Final.p df
61. Medicines and Healthcare products Regulatory Agency. Rules and Guidance for Pharmaceutical Manufacturers and Distributors 2017 (The Orange Guide). London: 2017.
62. Yang L, Sun G, Eppler MJ. Making strategy work: a literature review on the factors influencing strategy implementation [thesis]. Handbook of research on Strategy Process, 2010.
63. Khoja SS, Khoja SS, Chauhan PH, Khoja FS, Khoja SS. A review on creation and handling of data in accordance with cGMP requirements in pharmaceuticals. PharmaTutor. 2017;5:64-9.
64. Wingate G. Computer systems validation: quality assurance, risk management, and regulatory compliance for pharmaceutical and healthcare companies. Interpharm/CRC: Boca Raton, FL; 2004.
65. Craig R, Hickman CR, Silva MA. Creating excellence. 2018. doi:10.4324/9781351065306.
66. Guiso L, Sapienza P, Zingales L. The value of corporate culture. J Financ Econ. 2015;117:60-76.
67. Singh A, Joshi R. Analyzing the state of Data Integrity compliance in the Indian pharmaceutical industry. Ernst & Young LLP: India; 2015.
68. Bhatia S. Creating a culture of honesty and integrity in supply chains. Asian J Manag Sci Ed. 2018;7:55-61.
69. Alfred C. Maximizing leverage from an object database. IBM Systems Journal. 1994;33:280-99. doi:10.1147/sj.332.0280.
70. Nadkarni PM. Metadata-driven software systems in biomedicine. Health Informatics. London: Springer; 2011.
71. Schmidt J. How big data is transforming pharmaceutical manufacturing. Pharmaceutical Online. 2016.
72. Gandomi A, Haider M. Beyond the hype: big data concepts, methods, and analytics, Int J Inform Manage. 2015;35(2):13744.
73. Moniruzzaman ABM, Hossain SA. NoSQL Database: new era of databases for Big Data analytics – classification, characteristics and comparison. Int J Database Theory Appl. 2013;6(4):1-14.
74. Praveen S, Chandra U, Ali A. A Literature review on evolving database. Int J Comput Appl. 2017;162(9):35-41.
75. Gudivada VN, Rao D, Raghavan V. Renaissance in Data Management Systems: SQL, NoSQL, and NewSQL [thesis]. 2015.
76. Harrison G. Next generation databases: NoSQL, NewSQL, and Big Data, IOUG. Chicago; 2015.
77. Dove AP, Jundt LO, Lucas J. Using an object-oriented database as a repository for process configuration data. ISA Trans. 1997;36(4):339-43.
78. Haerder T, Reuter A. Principles of transaction-oriented database recovery. ACM Computing Surveys. 1983;15(4):287-317.
79. Jaedicke M, Mitschang B. User-defined table operators: enhancing extensibility for ORDBMS. VLDB’99, Proceedings of 25th International Conference on Very Large Data Bases. Edinburgh, Scotland, UK; 1999. p. 494-505.
80. Sahafizadeh E, Nematbakhsh MA. A survey on security issues in Big Data and NoSQL, Advances in Computer Science: an International Journal. 2015;4:69-72.
81. Bernstein D. Todays Tidbit: VoltDB. IEEE Cloud Computing. 2014;1(1):90-2.
82. Agarwal AK, Srivastava A. Study, comparison and analysis of different types of storage systems. Int J Curr Eng Tech. 2016;6(4):1392-9.
83. Hong W. Object-relational DBMS. 2015.
84. Ingolikar R, Khandal R, Mohare R. Comparison of HDBMS, NDBMS, RDBMS and OODBMS. Int J Adv Res Comp Sci Manage Studies. 2015;3(6):119-27.
85. Phaneendra VS, Reddy ME. Big Data – solutions for RDBMS problems – a survey, Int J Adv Res Comp Sc Software Eng. 2013;2:3686-91.
86. Agarwal AK, Srivastava A. Study, comparison and analysis of different types of storage systems, Int J Curr Eng Tech. 2016;6(4):1392-9.
87. Chandrasekhar R. Object Oriented DBMS (OODBMS). SQL Versity. 2014.
88. Corbellini A, Mateos C, Zunino A, Godoy D, Schiaffino S. Persisting big- data: the NoSQL landscape. Information Systems. 2017;63:1-23.
89. VoltDB, 4.2. Updating the Database Schema, 4.2. 2015.
90. Edlich S. Choose the “Right” Database and NewSQL: NoSQL Under Attack, InfoQ. 2012.
91. McCreary D, Kelly A. Making sense of NoSQL: a guide for managers and the rest of us. Manning: Shelter Island; 2014.
92. Haines S. Introduction to HBase, the NoSQL Database for Hadoop. Policy | InformIT. 2014.
93. Okman L, Gal-Oz N, Gonen Y, Gudes E, Abramov J. Security issues in NoSQL Databases. IEEE 10th International Conference on Trust, Security and Privacy in Computing and Communications. 2011. p. 541-7.
94. Srinivasa KG, Hiriyannaiah S. Comparative study of different in-memory (No/New) SQL Databases, A deep dive into NoSQL Databases: the use cases and applications. Advances in Computers. 2018;109:133-156.
95. Laranjeiro N, Soydemir SN, Bernardino J. A survey on data quality: classifying poor data. 2015 IEEE 21st Pacific Rim International Symposium on Dependable Computing (PRDC). 2015.
96. Clark B, Burstall R. Blockchain, IP and the pharma industry—how distributed ledger technologies can help secure the pharma supply chain. J Intellectual Property Law & Practice. 2018;13:531-3.
97. Conoscenti M, Vetro A, Martin JCD. Blockchain for the Internet of things: a systematic literature review, 2016 IEEE/ACS 13th International Conference of Computer Systems and Applications (AICCSA). 2016.
98. Crosby M, Nachiappan, Pattanayak P, Verma S, Kalyanaraman V. Blockchain technology: beyond Bitcoin. Applied Innovation Review. 2016.
99. British Standards Institution. Deshpande A, Stewart K, Lepetit L, Gunashekar S. Overview report distributed ledger technologies/blockchain: challenges, opportunities and the prospects for standards. 2017 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.bsigroup.com/LocalFiles/zh-tw/InfoSec-newsletter/No201706/download/BSI_Blockchain_DLT_Web.pdf
100. Christidis K, Devetsikiotis M. Blockchains and smart contracts for the Internet of things. IEEE Access. 2016;4:2292-303.
101. Iansiti M, Lakhani KR, Mohamed H. The truth about blockchain, Harvard Business Review. 2017.
102. BlockchainHub. Blockchains & distributed ledger technologies. 2016 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://blockchainhub.net/blockchains-and-distributed- ledger-technologies-in-general/
103. Bocek T, Rodrigues BB, Strasser T, Stiller B. Blockchains everywhere – a use-case of blockchains in the pharma supply-chain. 2017 IFIP/IEEE Symposium on Integrated Network and Service Management (IM). 2017. p. 772-7.
104. Wentworth S. How will blockchain impact pharma? The Pharma Letter. 2018.
105. modum. Applications – Industry Specific, Modum. 2018 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://modum.io/solution/applications
106. Puthal D, Malik N, Mohanty SP. Everything you wanted to know about the blockchain. IEEE Consumer Electronics Magazine. 2018;7:6-14.
107. Autor D, Kaufman Z, Tetzlaff R, Gribbin M, Dole M, Glover D, et al. PDA points to consider: best practices for document/data management and control and preparing for Data Integrity inspections. PDA J Pharma Sci Technol. 2018;72:332-7.
108. Yli-Huumo J, Ko D, Choi S, Park S, Smolander K. Where is current research on blockchain technology?—a systematic review. Plos One. 2016;11.
109. Watanabe H, Fujimura S, Nakadaira A, Miyazaki Y, Akutsu A, Kishigami JJ. Blockchain contract: a complete consensus using blockchain. 2015 IEEE 4th Global Conference on Consumer Electronics (GCCE). 2015. p. 577-8.
110. Liu Y, Lu Q, Xu X, Zhu L, Yao H. Applying design patterns in Smart contracts. In: Chen S, Wang H, Zhang L.-J. (editors). Blockchain – ICBC 2018: First International Conference, Held as Part of the Services Conference Federation, SCF 2018 Seattle, WA, USA, 25-30 June 2018, Proceedings. Springer International Publishing, Cham. 2018. p. 92-106.
111. McDowall RD. Validation of chromatography data systems: ensuring data integrity, meeting business and regulatory requirements. The Royal Society of Chemistry. Cambridge; 2017.
112. Barkow S, Takahashi K. Current expectations and guidance, including data integrity and compliance with cGMP. Society of Quality Assurance Annual Meeting 2017. Available from: https://scholar.google.com.sg/scholar?q=Barl ow+S,+Takahashi+K,+Current+expectations+and+guidance,+including&hl=e n&as_sdt=0&as_vis=1&oi=scholart
113. Albon KI, Davis D, Brooks JL. A risk-based approach to Data Integrity. Pharma Technol. 2015;39(7):46-50.
114. Unger B. Is GMP quality system auditing fundamentally flawed? – a Data Integrity alternative. Pharmaceutical Online. 2017.
115. Morris J. Practical data migration. Bcs Learning & Development Lim. 2012.
116. Warmuth A, Dufrasne B, Appel J, Bauer W, Douglass S, Klee P, et al. Ds8870 data migration techniques. IBM Redbooks. 2015.
117. Wolfgang W, Ludwig H. Part 3: Ensuring data integrity in electronic records. BioPharm. 2000;13(11):S24-S27.
118. McDowall RD. What is Data Integrity training?, Spectroscopy. 2015;30:34-41.
119. ECA Certified Data Integrity Manager. ECA Academy.
120. Data Integrity Pharmaceutical Training Course, RSSL. 2020. http://rssl.cms.mdlzapps.com/pharmaceutical-training/pharmaceutical-training-courses/data-Integrity-and-electronic-records-and-signatures
121. Vaittinen S. Supply chain management in a highly regulated environment – a case study of supplier GMP-compliance management in the pharmaceutical industry [thesis]. Aalto University; 2016.
122. Maguire J, Peng D. How to identify critical quality attributes and critical process parameters. 2nd FDA/PQRI Conference on Advancing Product Quality; 5-7 October 2015; North Bethesda, Maryland
123. Rx-360. Rx-360 Best Practices Quality Agreement Guide. 2015. Available from: https://www.a3p.org/rx-360-best-practices-quality-agreement-guide/
124. Lopez O. Data Integrity expectations of EU GMP inspectors. PharmTech. 2017;41(7):6-10.
125. Lazar MS. “GMP” an evolution in application. Journal of GXP Compliance. 2008;12(2):20-3. Available from: https://go.gale.com/ps/anonymous?id=GALE%7CA175286944&sid=googleScholar&v=2.1&it=r&linkaccess=abs&issn=15525791&p=AONE&sw=w
126. Pharma Quality Europe, EU and US Joint Inspections: Data as the Cornerstone for the Future, FDAnews’ 8th FDA Inspections Summit. 2013. Available from: https://www.fdanews.com/ext/resources/files/Conference/FIS13Presentations/DIncerti–EU-and-US-Joint-Inspections-Data-as-The-Cornerstone-for-The-Future.pdf
127. Sawant A, Tetzlaff R, Baker D. 2018 PDA/FDA Joint Regulatory Conference; 9 July 2018. PDA Letter. 2018. Available from: https://www.pda.org/pda-letter-portal/columns/regulation-snapshot#di-code
128. Milmo S. Harmonizing GMP inspections. PharmTech. 2018;42(5):6-8.
129. ASQ. What is auditing? ASQ. 2015. Available from: https://asq.org/quality-resources/auditing
130. Pharmaceutical Inspection Convention, Pharmaceutical Inspection Co-operation Scheme. Guide to good manufacturing practice for medicinal products (Part 1). 2009 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://www.tga.gov.au/sites/default/files/manuf-pics-gmp-medicines-part1.pdf
131. International Society for Pharmaceutical Engineering. GAMP 5 guide: compliant GxP computerized systems. 2008 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://ispe.org/publications/guidance-documents/gamp-5
132. van Rijmenam M, Erekhinskaya T, Schweitzer J, Williams M-A. Avoid being the Turkey: how big data analytics changes the game of strategy in times of ambiguity and uncertainty. Long Range Planning. 2018;52(5):
133. McDowall RD. What’s new with the FDA’s Data Integrity guidance? Spectroscopy. 2016;31:1-6.
134. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Pharmaceutical Development Q8(R2). 2009 [homepage on the Internet]. [cited 2020 Sep 15]. Available from: https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf
135. Coronel C, Morris S. 1-3a – Role and Advantages of the DBMS. Database systems: design, implementation and management. 12th ed. 2016. p. 6-11.
|
Author for correspondence: Adjunct Associate Professor Sia Chong Hock, BSc (Pharm), MSc, Senior Consultant (Audit and Licensing) and Director (Quality Assurance), Health Products Regulation Group, Health Sciences Authority Singapore, 11 Biopolis Way, #11-01 Helios, Singapore 138667 |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


