2nd MENA Stakeholder Meeting on Biosimilars 2018 – Report
Published on 06 August 2019
Generics and Biosimilars Initiative Journal (GaBI Journal). 2019;8(2):76-87.
|
Introduction: The number of biosimilars, or similar biotherapeutic products, is expanding rapidly worldwide. A further 12 blockbuster biologicals are set to add to this number by 2020. Agreement about the best practices for the regulation, use, interchangeability and pharmacovigilance of biosimilars is lacking in many countries including in the Middle East and North Africa (MENA). |
Introduction
Biosimilars, or similar biotherapeutic products, have the potential to secure global supply of high quality, safe biological medicines while also cutting healthcare costs. To achieve this, several barriers need to be overcome. First, an internationally agreed system of regulatory approval will be necessary. Second, effective pharmacovigilance of biosimilars – via a system that can be accessed globally – is required. Third, a global understanding of and agreement on interchangeability must be worked out.
Countries within the Middle East and North Africa (MENA) region face a growing demand for the management of chronic diseases for which biosimilar treatments exist – including diabetes (for which biosimilar insulin can be used) and cancer (for which biosimilar rituximab and trastuzumab exist). According to His Excellency Dr Amin Hussain Al Amiri of the United Arab Emirates (UAE) Ministry of Health and Prevention (MOHAP), the MENA region has the highest prevalence of diabetes in the world, with more than one in 10 adults now living with the condition. According to the World Health Organization (WHO), cancer is the second leading cause of death globally after heart disease, being responsible for an estimated 9.6 million deaths in 2018 [1].
To achieve global agreement on regulatory approval, pharmacovigilance and interchangeability of biosimilars, open discussion across national borders and between different stakeholder groups is key. The opinion of one stakeholder group will often influence the opinion of another, and the opinion of one group is no more or less important than the opinion of another. A recent study looking at patient-reported outcomes (PROs) in separate trials of a biosimilar and originator insulin found no statistically significant differences in PRO between the biosimilar and the originator. The authors suggest that such outcomes should build confidence not only for patients but also for their healthcare providers in use of biosimilar insulins [2].
The number of biosimilars being used, or soon to be used, in MENA countries continues to grow as it does worldwide. Twelve biological products with global sales of more than US$67 billion will be exposed to biosimilar competition by 2020 [3], allowing patients access to affordable biological therapies for oncology, haematology, dermatology and autoimmune diseases.
Differences remain around the issues of regulatory approval, pharmacovigilance and interchangeability of biosimilars across the MENA region. The Generics and Biosimilars Initiative (GaBI) invited participants from countries in the MENA region and beyond including practising healthcare professionals, regulators and pharmaceutical manufacturers to discuss these issues. The goal of the meeting was to have all attendees, not just speakers, identify areas of consensus about how biosimilars should be regulated, used and monitored, as well as how to communicate the biosimilars concept to physicians, patients and other interested parties. The meeting also included case studies selected to highlight the importance of structure-function relationships for biosimilar monoclonal antibodies (mAbs), and how these relate to quality control and clinical performance.
Methods
On 10 October 2018, GaBI held the 2nd MENA Stakeholder Meeting on Regulatory Approval, Clinical Settings, Interchangeability and Pharmacovigilance of Biosimilars. As with previous workshops [4, 5], the objective of the meeting was to review and explain how biosimilars are currently regulated, used and monitored and to identify areas of consensus. In addition to presentations by expert speakers from Canada, Europe, the MENA region and the US, participants were instructed to discuss selected case studies on biosimilar mAbs in order to interpret the importance of structure-function relationships and understand their impact on quality control and clinical performance.
Results
Expert speaker presentations
Regulatory standards and practices on biosimilars in UAE: safety and efficacy
Dr Rasha Sayed Salama, Public Health Consultant and Public Health Advisor for Public Health Policy and Licensing Sector at the MOHAP UAE, opened her presentation with the aim of UAE’s ‘National Strategy for Innovation’ to make UAE one of the most innovative nations in the world within seven years and within seven sectors, one of which is health. Quoting the country’s Vice President and Prime Minister, Sheikh Mohammed bin Rashid Al Maktoum, she noted that ‘change creates great opportunities, renews ideas and forces everyone to think in a different way’.
Biopharmaceuticals are one of the fastest-growing segments of the pharmaceutical industry and are expected to represent over 50% of the market by 2020. The pressure to reduce healthcare expenditure while increasing patient access to these treatments will drive the development of biosimilars. Biosimilars represent a paradigm shift in the way safety and efficacy are assessed, and education – involving academic research, policymakers and patients values – is clearly needed to support their introduction.
Referring to biosimilar guidelines around the world – from the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human use (CHMP) Guideline on similar biological medicinal products [6, 7] to the US Food and Drug Administration’s (FDA) Abbreviated Approval Pathway for Biosimilars [8] – Dr Salama introduced the UAE requirements for registration of biosimilar products. These include data on consistency in the manufacturing process; heterogeneity assessment; therapeutic equivalence; safety and efficacy studies; demonstration of immunogenicity; and a pharmacovigilance plan where every person in the community can upload comments to the Ministry of Health directly.
In the UAE, biosimilars produced for overseas markets must follow international guidelines such as the US FDA and the European Union’s (EU) EMA. For national markets, biosimilars must follow UAE standards and guidance from the Cooperation Council for the Arab States of the Gulf (previously the Gulf Cooperation Council, GCC).
Any change in a biosimilar product’s specifications or characteristics is considered as a new product and will follow biosimilar procedures, see Figure 1.

Regulation of biosimilars in the EU – immunogenicity
Biosimilars are now firmly established in the EU as copy biologicals with a clear and effective regulatory route for approval, allowing for marketing of safe and efficacious biosimilar products. The EU was the first regulatory body to work on biosimilars, and thus has approved the most biosimilar products. Testing for unwanted immunogenicity is integral to product development, explained Dr Robin Thorpe, former Head of Biotherapeutics Group, National Institute for Biological Standards and Control, UK.
A biosimilar should be highly similar to the reference medicinal product in physicochemical and biological terms. Differences between originator and biosimilar versions (or, as Dr Thorpe prefers to call them, copies) are likely, and these need to be justified with regard to any potential impact on safety and efficacy. Importantly, differences are not a problem specific to biosimilars – this is an issue for biologicals in general. Testing for unwanted immunogenicity is integral to product development during both the clinical and post-marketing phases [9, 10].
Biological products (originators and bio-similars) can sometimes induce antibodies with different characteristics, including non-neutralizing antibodies against active (or inactive) product-related substances, binding antibodies against contaminants, neutralizing antibodies, or any combination of these. The clinical consequences of these vary, from benign to life-threatening.
Dr Thorpe cited an example where 13 out of 325 healthy platelet donors developed anti-platelet antibodies and transfusion dependence during treatment with megakaryocyte growth and development factor (MGDF). Only four out of 650 cancer patients treated with MGDF developed such antibodies. The reasons for this difference remain unclear, but are probably both product and patient related. The problem of induction of erythropoietin antibodies following treatment with certain EPO products and subsequent development of pure red cell aplasia was also mentioned [11]. Unwanted immunogenicity is impossible to predict, which is why immunogenicity studies are an essential component of clinical trials. Immunogenicity testing normally takes a tiered approach, including screening assays such as ELISAs and radioimmunoprecipitation assays, assays for confirming and assaying the specificity of antibodies, and neutralizing assays for discriminating neutralizing and non-neutralizing antibodies, see Figure 2. If the immunogenicity profiles of originator and biosimilar products are significantly different, they can be considered dissimilar.

Comparative immunogenicity studies – between originator and biosimilar products – need to be designed to demonstrate whether the immunogenicity of the products is the same or significantly different. For this, a homogenous and clinically relevant patient population should be selected, with head-to-head studies, using the same assays and sampling strategies. Post-approval assessment, usually as part of pharmacovigilance surveillance, is normally needed.
Biosimilars regulatory considerations in Saudi Arabia
Professor Aws Alshamsan discussed the regulation of biosimilars in Saudi Arabia by comparing the current system of department-centric evaluation for biosimilar approval – where each department separately approves each new therapy – with an alternative product-centric evaluation – where the departments work together in sequence to approve a therapy, see Figure 3. Professor Alshamsan says the process of department-centric approval is a disconnected, inefficient process taking 290 days, whereas product-centric approval takes much less time. Currently, only five biosimilars are approved in Saudi Arabia (Omnitrope, Remsima, Zarzio, Grastofil and Binocrit), compared with at least 50 approved in the EU [12].

Biosimilars are, on average, 37% cheaper than originator products, driving the need for a more efficient, faster, but no less safe and effective route to biosimilar approval. A product-centric evaluation would be considerably faster and more efficient, cutting approval time significantly, see Figure 3. Learnings from the product-centric system would help build internal capacity for the regulator.
Currently, changing from an innovator drug to its biosimilar, or changing between biosimilars, is acceptable in Saudi Arabia following discussion between physician and patient. Pharmacists in Saudi Arabia cannot substitute biosimilars without consultation with treating physicians. Interchangeability is approved at the regulator level; switching is approved by the local authority, prescriber and the patient; substitution is approved by the local authority and pharmacist. To date, no interchangeable biosimilars have been approved in Saudi Arabia.
Biosimilars: Canada’s approach to inter-changeability, biosimilarity, extrapolation of indications and uses – a comparison to the US FDA
Dr Jian Wang, Division Manager at the Centre for Evaluation of Radiopharmaceuticals and Biotherapeutics, Biologics and Genetic Therapies Directorate, Health Canada (HC), compared the FDA’s approach to biosimilars with that taken by Canada. In the US, biosimilars follow a biosimilar specific regulatory pathway – 351(K) – whereas in Canada, biosimilars are considered as new drugs. In Canada, a stepwise approach to biosimilar approval progresses from comparative quality studies, to comparative non-clinical studies, to comparative clinical studies, see Figure 4. FDA compares biosimilars against US-licensed reference products, whereas the use of a non-Canadian version of the Canadian authorized reference product is acceptable by HC. Bridging to the national product is not required by HC, whereas FDA does require bridging to demonstrate that any foreign-sourced reference product and the corresponding domestic product are highly similar [13].

Looking at pharmacokinetic (PK) endpoint acceptance limits, FDA considers that the 90% confidence interval (CI) of the relative mean maximum concentration (Cmax), area under the serum concentration–time curve (AUCt) and area under the curve with respect to increase (AUCi) of the test to the reference should be within 80% to 125%. HC considers that the 90% CI of the relative mean AUCt and the 90% ratio Cmax of the test to the reference should be within 80% to 125%. With the pharmacodynamic (PD) endpoint, FDA considers that the 90% CI, for mean ratio (test to reference) should be within the predefined acceptance limits of 80% to 125%. HC considers that the 95% CI, for mean ratio (test to reference) should be within the predefined acceptance limits of 80% to 125%.
Dr Wang identified several potential differences in clinical trial design (in oncology) between the US and Canada, see Table 1, including differences in statistical analysis on endpoint (risk ratio for FDA; risk difference for HC), and in study endpoint (pathological complete response (pCR) for FDA, HC also accepts pCR, even though pCR-based indication has not been authorized in Canada and overall response rate (ORR) is preferred.

Interchangeability
On the issue of interchangeability, HC’s authorization of a biosimilar is not a declaration of equivalence to the reference drug, and the authority to declare two products interchangeable rests with each province and territory. At FDA, interchangeability designation and standards are mandated by law (Final guidance published in May 2019), and there are additional data requirements.
FDA has different and distinct statutory approval requirements for biosimilars compared with interchangeable (IC) products. Additional data are required for IC products: they are expected to produce the same clinical result in any given patient; and any risk in terms of safety or diminished efficacy of alternating or switching between IC and reference products should be no greater than the risk of continuing with the reference product alone. It is the applicant’s responsibility to choose their approach; and provide adequate support for their approach in addressing these additional requirements.
At the time of this meeting (10 October 2018), no interchangeable biosimilar product had been licensed by FDA [14].
No switch study
On the issue of switching, HC does not require a switch study from a reference biological drug to a biosimilar for market authorization.
When HC and relevant regulatory agencies authorize the marketing of a biosimilar, that biosimilar will have met all quality, safety and clinical standards. The final authorized indications are not extrapolated from one single comparative clinical study. The decision to authorize the requested indications is dependent on the demonstration of similarity between the biosimilar and reference biological drug based on data from comparative structural, functional, non-clinical, PK/PD and clinical studies and a detailed scientific rationale.
The approved biosimilar will be structurally and functionally (highly) similar to the reference product. Residual uncertainty from quality assessment does not cause clinically meaningful differences in efficacy, safety and/or immunogenicity. A biosimilar approved by HC may receive all or some therapeutic indications of the reference product.
Biosimilars receive indications of the reference based on the totality of evidence collected from all comparative studies, see Figure 5.

Principles and challenges related to manufacturing process development and demonstration of analytical comparability for biosimilars
The concept of analytical similarity, from the EU biosimilar perspective, was discussed by Dr Niklas Ekman, Vice Chair of the EMA’s Biosimilar Medicinal Products Working Party (BMWP). He looked at this from development of the manufacturing process for biosimilars, and from product experience, reflecting on two recent marketing authorization application assessments (Remsima/Inflectra infliximab; and Ontruzant trastuzumab).
The development programmes for biosimilars start with a comprehensive characterization of the chosen reference medicinal product, the Quality Target Product Profile (QTPP). This requires extensive analytical testing, including physicochemical and biological assays. The biosimilar manufacturer aims to achieve a deep understanding of the quality profile of the reference product, including the naturally occurring batch-to-batch variability, see Figure 6.

The characterization data also guides the development of the manufacturing process for the biosimilar. Once the process has been established, the pivotal evidence of analytical similarity is obtained through characterization studies which should, as far as possible, be conducted in a side-by-side manner. The comparison should include a sufficient number of biosimilar and reference batches using orthogonal, highly sensitive and appropriately qualified analytical methods that are able to detect all relevant differences in the quality profiles. When properly completed, the analytical similarity exercise significantly reduces the remaining uncertainty with regard to biosimilarity.
The experience of the EMA CHMP in assessing biosimilarity was highlighted by the examples of two cases, one of which addressed the impact of glycosylation differences observed for Remsima/Inflectra infliximab; the other of which focussed on challenges associated with a shift in critical quality attributes of the reference product, Herceptin.
In the first case, high similarity was shown between the biosimilar Remsima/Inflectra and the reference (Remicade) for primary, secondary and tertiary structure, in vitro tumour necrosis factor-alpha (TNF-α) neutralization, binding affinity and in vitro functional tests. However, minor differences were reported for C-terminal lysine content, aggregates, intact IgG level, charged molecular variants, glycosylation pattern, and binding to FcγRIIIa.
Largely based on the results from additional functional in vitro studies, the CHMP concluded that the differences were not clinically meaningful. Functional difference was seen only in an antibody-dependent cell-mediated cytotoxicity (ADCC) assay employing artificially high transmembrane ™ TNF-α expressing Jurkat target cells in combination with highly purified natural killer (NK) effector cells. Furthermore, there were no published reports of ADCC being induced by TNF antagonists in patients.
In the second case, analytical and functio-nal similarity were demonstrated between Ontruzant and EU Herceptin. However, in extended clinical characterization studies, comparability within the Herceptin originator could not be demonstrated using equivalence testing. As a result, the biosimilar had effectively become superior to the reference product, see Table 2.

Such differences raise important issues related to safety, but in this case the originator and biosimilar were shown to have similar safety profiles. Ontruzant was considered highly similar to Herceptin, and any differences in efficacy have since been attributed to a change in batches of the reference product.
Quality shifts of the reference product do happen, notes Dr Ekman. These can be challenging to handle for the biosimilar developer. Without reference product characterization data spanning over a long period of time, quality shifts might remain undetected. A thorough understanding of both the candidate biosimilar and the reference product characteristics is critical for successful biosimilar development, see Box 1.
Clinical and non-clinical assessment of biosimilars
Professor Andrea Laslop, Head of the scientific office at the Austrian Agency for Health and Food Safety, Austria, summarized the stepwise approach to demonstrating comparability between biosimilars and originators. After having shown biosimilarity at the physicochemical, biological and in vitro functional level, the clinical comparability exercise establishes biosimilarity in vivo.
If differences are observed at the non-clinical level, it is important to ask whether there is a plausible rationale (based on the quality characterization). It is important to ascertain whether the differences concern a major pathway or mode of action, and to consider whether further investigations using either more sensitive assays or under more physiological conditions would be useful.
All non-clinical findings need to be interpreted in the context of clinical results and vice versa – this is the principle of the totality of data.
PK similarity to the reference product is investigated in comparative phase I studies, mainly in healthy volunteers. These studies also yield comparative PD, supporting the evidence for biosimilar efficacy with limited information.
For PK/PD results outside equivalence limits, this might be because the study was not sufficiently powered, because the variability was higher than expected or because the drug concentration was higher in the test/reference batches. Variability can be difficult to anticipate if PK data on the originator product is scarce. Variability is also impacted by sample size. For these reasons, a crossover design may reduce or even eliminate variability, a further decrease in variability might also be seen by using only male subjects.
A clinical phase III trial confirming equivalence in efficacy completes the clinical comparability exercise and provides safety data pre-approval. When such an efficacy trial is performed, limited PK/PD sampling in the patient population can qualitatively confirm the results observed in healthy volunteers and at the same time allows the assessment of PK after repeat administration. In certain instances, when a validated PD surrogate marker is available, the PK/PD study may suffice to confirm similar efficacy. Considering the low sensitivity of the clinical phase III trial in detecting potential differences between the products, its importance may be questioned. Likewise, waiving of the phase III study could enable the development of biosimilar orphan drugs by mitigating feasibility constraints due to small population sizes.
If differences in efficacy are seen, it is important to ask whether the study was sufficiently powered. Similarly, if the variability was higher than expected, this could potentially be addressed by providing additional PD data, in case these data would represent established surrogate markers of efficacy. Before investigating further, it should be determined whether the observed differences are clinically relevant, as usually defined by the chosen non-inferiority margin.
If differences in safety are observed it is important to check that these were not just a chance finding. They could be the result of differences in antigenicity or in impurities, in which case a stringent risk management plan should be proposed in order to especially control the development of antibodies. However, differences in immunogenicity could also be the result of an artefact due to assay variability or due to a difference in assay sensitivity. Above all, strong post-marketing collection of safety and immunogenicity data is paramount.
Once biosimilarity has been established in one indication, extrapolation to other indications is generally accepted on the basis of sound scientific justification. Extrapolation is the most important principle for biosimilars, but it is also the most contentious. It is not a new concept – extrapolation is routinely seen with generics, for example, and in paediatric indications where trials on child subjects are often not deemed ethical. Another situation most relevant for the understanding of extrapolation for biosimilars is a change in manufacturing of an approved biological product. This leads to a new version of the active substance, which also corresponds to the definition of a biosimilar, indicating that in fact an originator biological after changes in manufacturing can be regarded as its own biosimilar. However, clinical data are typically not required to substantiate manufacturing changes and extrapolation is done on the basis of comprehensive in vitro comparison of the product before and after the manufacturing change. In any case, extrapolation should always be seen in the light of the totality of data.
An update on biosimilars and switching experience – the clinical perspective
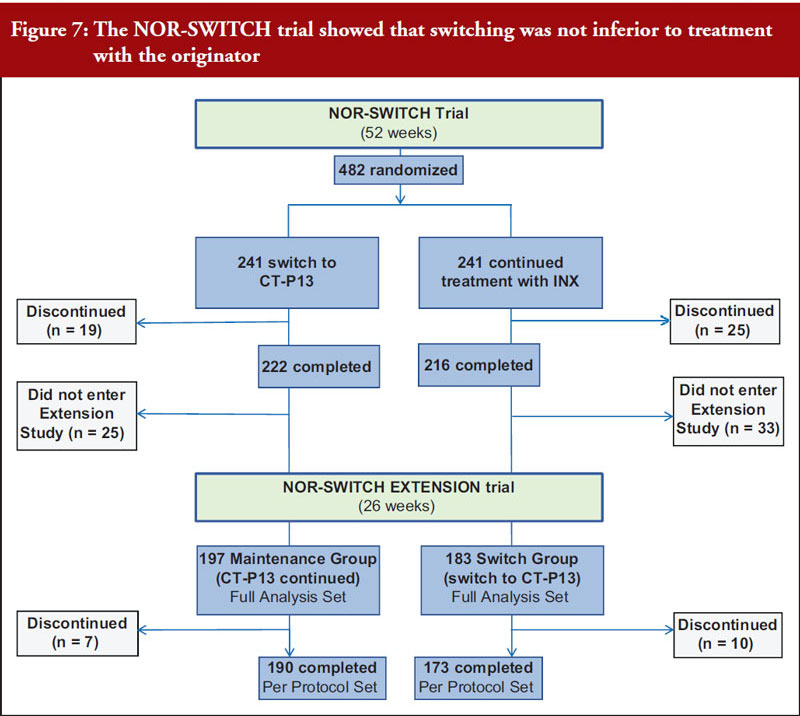
Professor Tore Kristian Kvien, who leads the rheumatology department at Diakonhjemmet Hospital in Norway, provided an update on the Norwegian government-funded NOR-SWITCH trial [15]. The trial involved 482 patients on stable treatment with Remicade across six indications including rheumatoid arthritis, Crohn’s disease, ulcerative colitis, spondyloarthritis, psoriatic arthritis and psoriasis. Patients were randomized to continue with the reference product or switch to Remsima/Inflectra and there were no significant differences in primary (disease worsening) or secondary (including drug discontinuation and remissions rates) endpoints. An extension study showed that switching from reference product to CT-P13 was not inferior to maintained treatment with CT-P13 over 26 additional weeks [16], see Figure 7. Professor Kvien concluded that these data add to the increasing real-world evidence that switching from originator biological disease modifying antirheumatic drugs (bDMARDs) to their biosimilars is safe and efficacious.
Despite this, Professor Kvien noted persistent objections to biosimilars based on subjective complaints – the Nocebo effect. This will need to be addressed in future studies [17].
Regulatory Panel Discussion
Professor Aws Alshamsan introduced the Saudi approach to interchangeability and switching by explaining that biosimilars are not automatically interchangeable. Of the 10 biosimilars that have been approved in Saudi, only two are deemed interchangeable. Biosimilarity alone is not enough for substitution or switching – this requires another level of scrutiny.
This approach is quite different to that of Tunisia, as explained by Dr Sonia Sebai Ep Ben Amor. In Tunisia, medicines are procured by pharmacy centralized purchase (PCP). When the biosimilar of trastuzumab Hertraz was approved by the Biosimilar Specialized Committee, the Minister of Health’s tender committee and the national insurance company chose to purchase the biosimilar for economic reason, which meant Herceptin was no longer available. There was no choice. Since then, a second biosimilar has been chosen and patients have been switched to this. Patients who prefer to get the originator have to pay for it themselves. Ongoing pharmacovigilance is the only way to record any differences in safety and efficacy between biosimilars and originator.
Tunisia’s Biosimilar Specialised Committee makes decisions on interchangeability on a case-by-case basis. The committee includes representatives of regulatory authorities, national control laboratory (dossier assessors), pharmaceutical inspection and the purchasing department as well as expert clinicians from different specialities who use biosimilars. There are several brands of insulin available in Tunisia, from Sanofi, Novo Nordisk and Julphar (Juslin). Two Tunisian insulins – human insulin and insulin glargin – are currently going through registration.
No monoclonal antibody biosimilars are currently marketed in Egypt, but two are under discussion, according to Ms Doaa Mohamed Abdelrady Mohamed. Decisions regarding interchangeability will be made by the Ministry of Health, and the patient will not be given a choice, said Ms Mohamed.
Dr Niklas Ekman summarized Finland’s, and more broadly the EU’s, approach. He highlighted issues around terminology. In the EU, interchangeability describes a property of two products, that are expected to gain the same result in the patient. Interchangeability can be seen in two ways: either as switching by the prescriber; or as substitution at the pharmacy level, without the knowledge of the prescriber. In Finland, all biosimilars that have been approved in the EU are interchangeable, which means that the physician can change between the two. Finland has not yet taken a position on substitution at the pharmacy level, but from a scientific point of view, the challenges of substitution should be similar to those of switching. There are, however, still practical questions associated with pharmacy-level substitution that need to be solved. EMA approves biosimilars, but Member States can decide whether to approve interchangeability. Most Member States agree that EMA-approved biosimilars are interchangeable when the switch is overseen by the prescriber.
Ms Dunia Ama AlBastaki, who works in drug regulation in Kuwait, asked whether approved biosimilars were themselves interchangeable. Dr Ibrahim A Aljafalli, former Vice President of the Drug Sector in Saudi FDA, replied that biosimilars approved by EMA were considered inter-changeable.
In response to a question from Ms Suna Mohammad Ibraheem Habahbeh, Professor Alshamsan explained that, in order to approve switching, a clinical trial that involves switching must be run. This must happen before the biosimilar is approved.
Professor Ahmed Elmelegy of Egypt asked about the extent of biosimilar price differences in Saudi and Tunisia, Dr Sebai Ep Ben Amor of Tunisia said that there was a 50% difference between Herceptin and its biosimilar – an enormous reduction. Professor Alshamsan said that there was currently no price difference between Remicade and Remsima, but for Neupogen and Zarxio the biosimilar was a fifth of the price. He expected a new pricing policy would be introduced soon. Professor Abdalla Mohamed Elsayed Abotaleb of Egypt added that Egypt has a pricing policy for biosimilars based on a concept called second brand innovator. There is a 30% variation from innovator to second brand pricing for Hervive and Retuksera.
Summary of the discussions that followed the expert presentations
Regulatory standards
Mazen Kurdi, Professor of Pharmacology at the Lebanese University, argued that most regulatory bodies in the MENA region are made up of pharmacists, with a few physicians. Based on the information shared at this meeting, Professor Kurdi said it was clear that regulatory bodies need scientists. The science is not always understood by regulatory bodies, and this is particularly a problem for locally-produced drugs (rather than those imported from the US and Europe, which have been subject to more informed regulation).
Dr Aljafalli, responded that Professor Kurdi’s generalization was not fair to mature regulatory bodies in the region. Many regulatory bodies in the region have experts other than pharmacists. In the Saudi FDA, for example, there are chemists, biochemists, microbiologists, analytical chemists, biostatisticians, pharmacists, physicians and experts in veterinary medicine. Dr Aljafalli, a pharmacist himself, said many regulatory authorities in the region were similarly diverse.
Dr Rasha Sayed Salama, UAE, said biosimilars produced in the UAE are approved by the country’s Public Health Policy and Licensing Sector, which has a Drug Department responsible for approval. This includes pharmacists, academics and members of the pharmaceutical sector. For applications from the EU and the US, EMA and FDA guidelines are followed (with some revisions).
Immunogenicity
Dr Robin Thorpe, UK, said that the latest immunogenicity guidance from EMA was issued at the end of 2017 but came into effect in 2018. There is currently no better guidance and it can only improve as fast as technology improves. Asked if improvements in post-approval assessment were also needed, Dr Thorpe replied that they were and warned that, particularly in developing nations, harmful side effects could be going undetected.
Dr Ekman, Finland, joined Dr Thorpe in explaining that immunogenicity is always tested in clinical studies. It is difficult to predict immunogenicity, but it is important to take into account features, for example, aggregated products, that might have the potential to be immunogenic. Dr Thorpe added that rheumatology patients are more ‘immune active’ and so need to be assessed as such.
Interchangeability and switching
Professor Alshamsan, King Saud University, Saudi Arabia, said that whereas guidance on interchangeability was the regulator’s responsibility, switchability is the decision of the prescriber. If two biosimilars are proven interchangeable, they need a switching study, he said. The regulator cannot force the prescriber to switch between biosimilars, even if they are interchangeable.
Biosimilars in Saudi Arabia
Professor Alshamsan said that two trastuzumab biosimilars are currently going through the registration process. Alongside these, there are registration plans for biosimilar rituximab and bevacizumab.
Biosimilar indications
Dr Jian Wang, Health Canada, explained that the number of indications a biosimilar has varies depending on the country. A biosimilar might have 10 indications in the US, but only eight in Canada. It cannot claim that it has 10 in Canada if the Canadian reference product only has eight.
Batch variability
Dr Ekman explained that EMA approves a product rather than batches. The approval process ensures consistent manufacturing, limiting batch-to-batch variation. Both Drs Ekman and Thorpe explained that the original approval is designed to take into account immunogenicity, usually including between six to 12 months of comparative immunogenicity data. The original approval should provide assurance, but pharmacovigilance is needed – as it is for all drugs.
Pharmacovigilance
Professor Andrea Laslop of the Austrian Agency for Health and Food Safety, responded to questions on pharmacovigilance in Europe, and explained how data are collected and assessed via periodic safety update reports (PSURs). Initially, these are based on a six-monthly interval and then once yearly and then, once the product has been on the market for over three years, not collected unless there is an issue with efficacy or safety.
Clinical trials
Although more extensive than a normal, quick and small PK trial, Professor Laslop said that crossover trials (where subjects receive each treatment in succession – acting as their own controls), are a worthwhile investment. Professor Laslop added that relatively large sample sizes – including PK studies with 180 to 190 healthy volunteers – were helpful when addressing high variability and increasing dropout rates.
Totality of evidence
A presentation on the totality of evidence illustrated the critical role of structure–function in biosimilar development using the example of monoclonal antibodies. The relationship between chemistry, manufacturing and controls (CMC) and clinical studies was examined. All biologicals, including biosimilars, have complex structures and multiple quality attributes. The relationship between product quality attributes and biological function is complex, as is the relationship between product quality attributes and process.
Stakeholder Panel Discussion
Clinical perspective on biosimilars use: clinicians, pharmacists, regulators
European and US approaches to pharmacovigilance of multisource biologicals were presented prior to a stakeholder panel discussion. When multiple sources of biologicals are available it is challenging to accurately trace and attribute adverse events (AEs) to the correct product. Misattribution of AEs can damage reference product safety databases and prevent biosimilar manufacturers from accurately measuring the benefit-risk of their products.
In Europe, via legislation and the release of good pharmacovigilance practice guidance, brand names and lot numbers are required to be recorded in patient medical records and AE reports. The intent is that all Member States will comply, and that accurate traceability will be achieved by efforts to improve reporting processes by users.
In the US, FDA has finalized a naming policy that adds a four-letter suffix to a common root non-proprietary name for all biologicals and biosimilars that are newly approved [18]. The intent is that a distinguishable naming convention will allow for automatic use on packaging and in systems during prescribing, dispensing, documentation in patient records, and when an AE is reported.
An effective system of pharmacovigilance for biosimilars from many different sources is key to fostering confidence in the biosimilar marketplace and to empowering manufacturers to take full accountability of their product profiles.
Biosimilar naming
Professor Aws Alshamsan questioned the value of the four-letter suffix. If a patient was receiving Neupogen, for instance, and then a week later was switched to Zarxio, how would an immunogenicity incident be reported? Would it be attributed to Zarxio or to Neupogen because the immune response took its time to build up, perhaps it was a delayed type of hypersensitivity. Professor Alshamsan wondered if the four-letter suffix would mislead the regulator to interpret the data incorrectly.
Dr Thomas Felix, Amgen, explained that a single report of an AE is not sufficient evidence to link an AE to a particular product. Each individual report is filed, and healthcare professionals are alerted when a signal or a cluster of reports becomes apparent in the records. A single report might go to the wrong place, but when an investigation is underway, the hyphenated suffix will help. Without the suffix you might only have the non-proprietary name. If you have a long history of different products, maybe cycling through three or four different biosimilars, you would notice over time how a particular product was linked to a particular event.
Dr Robin Thorpe, UK, added that Japan developed their biosimilar naming system with the prefix BS1, BS2, BS3 and so on before WHO developed its biological qualifier (BQ) system. FDA embarked on biosimilar naming much later and tried to base their system on some of the principles of the WHO BQ system, but this has created problems that Dr Thorpe does not see getting resolved.
Pharmacovigilance
Dr Mohamed A Omair from the rheumatology department at King Saud University, Saudi Arabia, agreed that pharmacovigilance is essential but also difficult to find time for alongside a busy physician’s workload. He suggested that hospital pharmacists should take the lead, particularly where there is a clinical pharmacy service.
Dr Omair cited a small study that found only 25% of Saudi rheumatologists thought biosimilars might have a role in patient management. A subsequent study during the Arab League Against Rheumatism meeting, which included Arab rheumatologists from across the Middle East region, asked physicians about their level of knowledge. Rheumatologists were asked whether they thought the evidence for biosimilars was adequate, whether they would use biosimilars, and how many years’ experience they would need before prescribing biosimilars to most of their patients. Thirty per cent of those questioned responded that there was insufficient evidence in favour of biosimilars, and they would not use them. About 30%−40% responded that they were unsure. Dr Omair and colleagues conclude that there is a clear knowledge gap among physicians. Education is needed, and this must focus on healthcare providers rather than decision makers and officials.
Switching
Dr Adeeba Al-Herz, President of the Kuwait Association of Rheumatology, based at the Amiri Hospital; Faisal and Medical One Specialized Polyclinic in Kuwait, raised the issue of difficulty in switching between known, effective biological originators and unknown biosimilars. Despite literature in support of a biosimilar, a physician is more familiar with the efficacy and safety of the originators given the fact that they have been using them for many years. In addition, physicians still need more data regarding their long-term safety. Patients will also be worried of taking unknown drugs, regardless of published evidence. Dr Jian Wang, from Health Canada, asked how much evidence would be needed to convince patients. Dr Al-Herz said that the evidence was sufficient, but evidence alone is not enough to convince patients (or even physicians) on a practical level. Dr Wang echoed Dr Niklas Ekman’s earlier presentation showing that there had been no examples – in the past 12 years – of an approved biosimilar behaving differently to its originator. Dr Ekman cautioned that this was true for the EU; but may be different in other regions. Dr Wang understood that patients would question why they were being switched when there was no problem with the originator: trust must be built between regulators, physicians and patients. Dr Wang suggested differences between originators and biosimilars might have been overemphasized, overlooking the high similarity between them.
Professor Tore Kristian Kvien emphasized the role of communication, and the importance in improving compliance. In Norway, the pressure to switch to a biosimilar is particularly great because of the cost differences, which are felt directly by hospital budgets. This press ure is not so marked in MENA countries.
Patient choice
Dr Ravi Mohan Pedapenki of the Ministry of Health in Bahrain, at the Salmaniya Medical Complex, Medical Oncology, was reminded of a patient in India treated successfully with an originator biological treatment for HER2+ (human epidermal growth factor receptor 2 positive) breast cancer (trastuzumab). When the patient’s insurance stopped covering the cost of treatment, the patient refused to switch to the cheaper biosimilar but could not afford the originator. Her symptoms returned. Finally, she had to be treated with the biosimilar, which was as successful as the originator had been. In this way both the physician and the patient were convinced of the efficacy of the biosimilar. Being provided with evidence by regulators is not enough, physicians and patients need to see evidence at first hand.
There was wide agreement from physicians across the MENA region that the economic argument in favour of biosimilars is off-putting. Switching from a known successful treatment to an unknown treatment because it is cheaper does not instil confidence, particularly if neither the patient nor the physician is under tight financial constraints.
In response to a question about how many times patients could be switched between biosimilars, Professor Tore Kristian Kvien, Norway, replied that a multi-switch trial had been designed and was waiting to receive funding. Professor Kvien says multi-switch trials are needed. It is not sensible to switch if disease symptoms are not being controlled. In this case it would be better to choose a therapy with a different mode of action.
Patient choice is key in Kuwait and Dr Yasser Mustafa Ali Ghadanfar from Kuwait predicts this will present a barrier to switching. Professor Kvien responded that patients in Norway were willing to switch to the biosimilar when they were told how much money would be saved, and how this could be used to offer more innovative therapies. Dr Wang agreed that the financial argument was the only way to convince patients – that improved treatments will be made available with the savings. Patients who still prefer not to switch have to pay for the difference for their own therapy.
Asked how much money was saved by switching to biosimilars in Norway, Professor Kvien said that in 2014 Remsima, biosimilar infliximab, was 39% less expensive than Remicade. In 2015, it was 69% less expensive. Pricing information is now publicly confidential, so Professor Kvien was unable to provide more recent figures.
Dr Wang, said savings were variable. The listed savings are between 15% to 50%.
Dr Mohammed A Omair, Saudi Arabia, mentioned a patient programme being designed to explain what biosimilars are and how they are used worldwide. This should improve patient acceptance of biosimilars and reduce the nocebo effect (caused by unwarranted negative expectations).
Case study on the importance of structure–function relationships for biologicals/biosimilars
Discussion groups were provided with data on two semi-fictional trastuzumab biosimilar candidates. They were provided with physicochemical characteristics, selected glycan and biological attributes, and the results of a phase I study for Candidates 1 and 2. Similar case study was carried out at previous GaBI meetings.
Each discussion group was asked whether the data for the candidates qualified for biosimilarity with a reference product from a quality (CMC) perspective. If not, they were asked what steps they would recommend fixing this. Discussion groups were also asked how ‘residual uncertainty’ could be addressed in preclinical or clinical studies. They were then asked, given that Candidates 1 and 2 had both the CDR (complementarity-determining region) and Fc (fragment crystallizable) region involved in their mechanism of action (MoA) for some of the indications, whether they would recommend extrapolation to all indications.
Summary discussion of case study of therapeutic protein monoclonal antibody – Candidates 1 and 2
Professor Ahmed Elmelegy from Egypt led discussion Group 1, concluding that they would approve Candidate 1 and not approve Candidate 2. Candidate 1 was deemed a biosimilar because its physicochemical characteristics – the MoA, the binding capacity and binding results – were similar. His group found no residual uncertainty and so did not require additional preclinical or clinical studies. The group agreed about extrapolation in all cases in which HER2 is expressed – breast cancer, metastatic breast cancer and gastric cancer. Candidate 2 was not biosimilar because the quality attributes were different. Importantly, the MoA, the binding to the HER2, was inferior. The clinical study results were also inferior. His team suggested the drug company would have to go back to the beginning with Candidate 2.
Professor Ahmed Aljedai of Saudi Arabia led Group 2; and agreed with the conclusions of the first group. However, Professor Aljedai’s group would have liked to see additional studies because there were some differences with regards to deamidation which might affect potency. The group would ask for a comparative non-clinical study and would recommend PD studies for this purpose. The group would address issues of residual uncertainty with these preclinical studies and with clinical studies. The group agreed that the study design was adequate for the given indication but they would require further studies before extrapolation to other indications.
Professor Laslop asked if Professor Aljedai’s group would recommend PD studies in patients. Professor Aljedai said they would start with animals and move to patients. They chose this way of making sure differences in deamidation were not affecting potency, otherwise they might be wasting time and money. Professor Laslop said that no PD studies would be recommended in Europe, particularly as animal studies would not give relevant information for human subjects.
Dr Ravi Mohan Pedapenki, Bahrain, said that he would treat all HER2+ cancer patients with Herceptin, regardless of the cancer type because the mechanism remains the same. Dr Jian Wang, who was in Professor Aljedai’s discussion Group 2, was concerned that the concept of biosimilarity was being overlooked by the group. Biosimilarity was accepted, but extrapolation was not recommended. Professor Aljedai asked who would extrapolate based on the MoA only or the totality of evidence including MoA as well as the physicochemical properties and others, without the need for clinical trials. If you take the example of rituximab, which has been studied in lymphoma patients, said Professor Aljedai, would you extrapolate to rheumatoid arthritis?
Dr Pedapenki said he would not, because he only extrapolated in cancers. Professor Laslop said that they do extrapolate in Europe. In Europe, they agree to the extrapolation for rituximab from rheumatoid arthritis to oncology and the other way around. Rituximab depletes CD-20 positive B cells, said Professor Laslop. That is the same PD effect, in each of the diseases where the originator has been approved, and this is why extrapolation works. She added that regulators would ask for an additional PK trial in the other population to check for differences in the PK profile. It could be different because in the oncology setting there is target mediated clearance which is not relevant for the immunological disease.
Group 2 was co-moderated by Professor Abdalla Mohamed Elsayed Abotaleb, Egypt, who explained that his group agreed with Group 1 that Candidate 2 did not qualify as biosimilar due to differences in physicochemical characteristics that might impact potency and immunogenicity. As before, the group said that this candidate would need to be started from scratch.
Group 3, moderated by Professor Mazen Kurdi, Lebanon, reported that his discussion group would ask for extra clinical data before extrapolating with Candidate 1 ‘even if it was clear that we should not’. He and his group concluded that this was needed because the candidate was a mAb and showed differences in deamidation. Dr Niklas Ekman commented that this would depend on where the deamidation was, adding that a clinical study might not reveal features relating to the deamidation. Deamidation differences will not always affect function.
Group 4, moderated by Dr Adeeba Al-Herz, Kuwait, concluded any differences in deamidation could be remediated by optimizing the manufacturing process, for example, by improving deamidation, which might increase potency. Her group would have preferred more data on ADCC (antibody-dependent cell-mediated cytotoxicity and CDC (complement dependent cytotoxicity) to close in on any differences in immunogenicity. Nevertheless, the group would recommend extrapolation to all indications for Candidate 1 because if it is effective in one indication it can be extrapolated to all, particularly since the CDC and ADCC are similar. Dr Al-Herz added, however, that since potency (deamidation) is affected by structure, more data would be needed for all indications. The group concluded that Candidate 2 would not qualify as a biosimilar due to marked differences and the physicochemical characteristics that might impact potency and immunogenicity.
Group 5, moderated by Professor Ali K Abu-Alfa, Head of the Division of Nephrology and Hypertension at the American University of Beirut, Lebanon, would be happy with extrapolation for Candidate 1 following a period of learning more about the drug. As before, Candidate 2 was not judged a biosimilar.
Dr Ekman asked Professor Laslop about the endpoints that should be used to determine biosimilarity for the two candidates. She replied that the ideal endpoint in early breast cancer would be pathologically complete response. This is very sensitive because it can be measured more accurately than all of the other endpoints.
Overall survival, especially in a setting with patients with different sizes and numbers of metastases, is influenced by many additional factors leading to a lot of background noise in the trial. As a result, you might see no difference and conclude on similarity simply because of the fact that the whole background noise is masking any minor differences in the efficacy outcome.
Conclusions
Delegates at the 2nd MENA Stakeholder Meeting agreed that they had learned a great deal; but cautioned that what they had learned needed to be made more widely available. ‘In terms of clinicians, I think their knowledge in biosimilars is not as good as you may expect’, said Dr Adeeba Al-Herz from Kuwait. ‘We need to step down and go to the clinicians and try to educate them about the different aspects of biosimilars’. Dr Ibrahim A Aljufalli agreed, comparing the current position of biosimilars with the introduction of generics in the 1970s and 1980s. As regulatory authorities improve alongside improved sensitivity in the detection of differences between products, and experience in switching grows, physicians and patients will grow to accept biosimilars and the savings this will mean for healthcare budgets.
Dr Niklas Ekman concluded by encouraging meeting attendees to return to the clinicians in their countries and explain what biosimilars are and what they are not. Once this information has been made widely available, clinicians can then decide whether or not to prescribe biosimilars.
Speaker Faculty and Moderators
Speakers
Respected Dr Lubna AlShaali, PhD, UAE
Professor Aws Alshamsan, BPharm, RPh, PhD, Saudi Arabia
Niklas Ekman, PhD, Finland
Thomas Felix, MD, USA
Professor Tore Kristian Kvien, MD, PhD, Norway
Professor Andrea Laslop, MD, Austria
Jennifer Liu, PhD, USA
Rasha Sayed Salama, MD, PhD, UAE
Robin Thorpe, PhD, FRCPath, UK
Jian Wang, MD, PhD, Canada
Moderators
Professor Abdalla Mohamed Elsayed Abotaleb, PhD, Egypt
Professor Ali K Abu-Alfa, MD, FASN, FAHA, Lebanon
Adeeba A A H Al-Herz, MD, FRCPC, FACP, Kuwait
Professor Ahmed Aljedai, PharmD, MBA, BCPS, FCCP, FAST, Saudi Arabia
Professor Ahmed Abdelsalam Mohamed Elmelegy, MSc, PhD, Egypt
Professor Siham Hamaz, MD, Morocco
Professor Mazen Kurdi, PhD, Lebanon
Doaa Mohamed Abdelrady Mohamed, MSc, Egypt
Mohamed A Omair, MD, Saudi Arabia
Ravi Mohan Pedapenki, MD, Bahrain
Sonia Sebai Ep Ben Amor, MD, Tunisia
Editor’s comment
Speakers and moderators had provided the discussion/conclusion of the group discussion, read the report and revised the content of the summary discussion.
Acknowledgement
The Generics and Biosimilars Initiative (GaBI) wishes to thank Dr Rasha Sayed Salama from the MOHAP UAE for her support to the organization of the meeting; the moderators in clarifying the information of the case study discussion when finalizing the meeting report; as well as Professor Andrea Laslop and Dr Robin Thorpe, Chair and Co-chair of the 2018 meeting, for their strong support through the offering of advice and information during the preparation of the meeting.
The authors would like to acknowledge the help of all the workshop speaker faculty and participants, each of whom contributed to the success of the workshop and the content of this report, as well as the support of the moderators and co-moderators in facilitating meaningful discussion during the parallel case study working sessions, presenting the discussion findings at the meeting, and contributing in the finalization of this meeting report.
Lastly, the authors wish to thank Dr Bea Perks, GaBI Journal Editor, in preparing and finalizing this meeting report manuscript and providing English editing support on the group summaries.
Competing interests: The workshop was sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
1. World Health Organization. Cancer [homepage on the Internet]. [cited 2019 Jul 25]. Available from: https://www.who.int/cancer/en/
2. DeLozier AM, Ilag LL, Perez-Nieves M, Kaushik P, Duan R, Pollom RK, et al. Patient-reported outcome measures in phase III trials of LY2963016 insulin glargine and reference insulin glargine products: ELEMENT 1 and ELEMENT 2. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):56-62. doi:10.5639/gabij.2018.0702
3. GaBI Online – Generics and Biosimilars Initiative. US$67 billion worth of biosimilar patents expiring before 2020 [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2019 Jul 25]. Available from: www.gabionline.net/Biosimilars/General/US-67-billion-worth-of-biosimilar-patents-expiring-before-2020
4. Aljedai AH, Alhomaidan AM, Trifirò G, Alshamsan AWs, Al-Foheidi M, Alsenaidy MA, et al. First GCC Stakeholder Meeting on Approval Process, Interchangeability/Substitution and Safety of Biosimilars 2017 – Report. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(4):158-63. doi:10.5639/gabij.2018.0704.032
5. Thorpe R, Griffiths E, Ekman N. First ASEAN educational workshop on regulation and approval of biosimilars/similar biotherapeutic products 2017 – Report. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(3):127-32. doi:10.5639/gabij.2018.0703.025
6. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 18 December 2014 [homepage on the Internet]. [cited 2019 Jul 25]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf
7. GaBI Online – Generics and Biosimilars Initiative. EU guidelines for biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2019 Jul 25]. Available from: www.gabionline.net/Guidelines/EU-guidelines-for-biosimilars
8. GaBI Online – Generics and Biosimilars Initiative. US guidelines for biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2019 Jul 25]. Available from www.gabionline.net/Guidelines/US-guidelines-for-biosimilars
9. European Medicines Agency. Immunogenicity assessment of biotechnology-derived therapeutic proteins [homepage on the Internet]. [cited 2019 Jul 25]. Available from: https://www.ema.europa.eu/immunogenicity-assessment-biotechnology-derived-therapeutic-proteins
10. European Medicines Agency. Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. 24 May 2012 [homepage on the Internet]. [cited 2019 Jul 25]. Available from: https://www.ema.europa.eu/documents/scientific-guideline/guideline-immunogenicity-assessment-monoclonal-antibodies-intended-vivo-clinical-use_en.pdf
11. Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469-75.
12. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2019 Jul 25]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
13. Kurki P. Potential changes to the FDA approach to biosimilars have a global impact. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):53-5. doi:10.5639/gabij.2018.0702.011
14. Niazi SK. Rationalizing FDA guidance on biosimilars—expediting approvals and acceptance. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):84-91. doi:10.5639/gabij.2018.0702.018
15. Perks B. Randomized non-inferiority trial fails to find inferiority switching from infliximab originator to CT-P13 biosimilar. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(4):188-9. doi:10.5639/gabij.2017.0604.042
16. Goll GL, Jørgensen KK, Sexton J, Olsen IC, Bolstad N, Haavardsholm EA, et al. Long-term efficacy and safety of biosimilar infliximab (CT-P13) after switching from originator infliximab: open-label extension of the NOR-SWITCH trial. J Intern Med. 2019;285(6):653-69.
17. Tweehuysen L, van den Bemt BJF, van Ingen IL, de Jong AJL, van der Laan WH, van den Hoogen FHJ, et al. Subjective complaints as the main reason for biosimilar discontinuation after open-label transition from reference infliximab to biosimilar infliximab. Arthritis Rheumatol. 2018;70(1):60-8.
18. GaBI Online – Generics and Biosimilars Initiative. FDA update on naming biologicals [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2019 Jul 25]. Available from: www.gabionline.net/Reports/FDA-update-on-naming-biologicals
|
Author for correspondence: Robin Thorpe, PhD, FRCPath, Deputy Editor-in-Chief, GaBI Journal |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2019 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



Thanks for the manuscript publication. I read with interest; the way it was presented should be an eye-opener to many countries in the MENA region who do not use Biosimilar/Generics!!!. Hope this article stimulates others to really think about using in many countries with fewer resources.
Best regards
Dr Ravi Mohan Pedapenki