Assessment of active pharmaceutical ingredients in drug registration procedures in Pakistan: implications for the future
Published on 2016/07/27
Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(4):156-63.
Author byline as per print journal: Babar Khan, BPharm, MPH, PhD; Brian Godman, BSc, PhD; Ayesha Babar, BPharm, MPhil; Shahzad Hussain, BPharm, MPhil; Sidra Mahmood, PharmD, MSc; Tahir Aqeel, BPharm, MPhil
|
Introduction: There are concerns over the quality of generic medicines in Pakistan. This is due to perceived non-compliance with good manufacturing practice (GMP), whereby the quality of the raw materials is not being assessed. If not addressed, this will impact on the potential for generics exports from Pakistan, as well as on patient care. Consequently, there is a need to assess the current assessment and regulatory situation in Pakistan and to recommend a way forward that ensures the future quality of products. |
Submitted: 7 July 2016; Revised: 8 November 2016; Accepted: 14 November 2016; Published online first: 25 November 2016
Introduction
The pharmaceutical market in Pakistan was worth approximately US$2.3 billion in 2014 [1]. At present, Pakistan produces a variety of medicines and meets approximately 90% of the demand for domestic finished products. However, currently Pakistan only produces a limited amount of the active pharmaceutical ingredients (APIs) needed for medicines consumed in Pakistan [2], with more than 90% of raw materials/APIs coming from China and India.
There are concerns over the quality of medicines manufactured in Pakistan due to perceived non-compliance with good manufacturing practice (GMP) requirements outlined by pharmaceutical manufacturers [3]. These include checks on the quality of APIs being used to produce oral tablets [3]. GMP requirements are included in the Drug (Licensing, Registering and Advertising) Rules (Schedule-B II), which were enacted in Pakistan in 1976 [4]. However, since then, GMP and other registration requirements have not been updated [5]. This means that there have been no updates or revisions following the creation of international standards or World Health Organization (WHO) guidelines [6]. With over 1,200 registered medicines and over 80,000 registered drug products in Pakistan [5], coupled with physician concerns over the safety and efficacy of lower cost generics, the country sees high levels of prescribing of originator products [5, 7, 8]. This is a public health concern as self-pay for medicines in Pakistan is widespread [9] and therefore there are implications for affordability and subsequent adherence rates, especially for treatment of chronic diseases affecting the lower paid [10–12]. Patient care is not compromised with generic medicines that meet agreed quality standards, including bioequivalence levels, across a wide range of disease areas [13–19]. Concerns over generic immunosuppressants are also reducing [20].
There is inconsistency between the information to be included in application dossiers required for authorizing a medicine in Pakistan by the Drugs (Licensing, Registering and Advertising) Rules, 1976, and those stated in the Drug Regulatory Authority of Pakistan (DRAP) Act, 2012 [21]. In this Act, Schedule-I states that the pharmaceutical dossier should include a set of the following documents for submission that give all information on the technical aspects of a product’s manufacture:
a. Master formula
b. All ingredients both active pharmaceutical ingredients and inactive excipients added with their safety profile data
c. Complete manufacturing procedure of the drug, biological or medical device
d. Quality control steps and procedures at each level of raw material selection, in-process testing, finished drug testing and stability testing
e. Clinical trial data and published reports about the safety and efficacy of the drug
f. Complete details of manufacturing plant and equipment, quality control laboratories and equipment
g. Warehouse capacities and facilities; details of human resources available and the latest cGMP (current good manufacturing practice) report shall also be part of this document set
h. Any other information required by the Registration Board for establishing the safety, efficacy, bioavailability, bioequivalence, or biosimilarity of the drug.
Section 7 (c) (ix) of the DRAP Act, also emphasizes the systematic implementation of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), WHO and US Food and Drug Administration (FDA) guidelines [3, 6, 22]. All of these guidelines suggest that application for the registration of medicines in a country should follow an internationally harmonized format known as the Common Technical Document (CTD). The CTD consists of five modules [23]:
- Module 1 Region-specific Administrative information
- Module 2 Quality overall summary (Overview and summary of modules 3 to 5)
- Module 3 Chemistry manufacturing and Controls (Quality)
- Module 4 Non-clinical/Preclinical (Safety)
- Module 5 Clinical (Efficacy)
However, despite many developing countries implementing such standards, these standards are not implemented in Pakistan. The lack of implementing such standards demonstrates the continued weaknesses of this registration process [5].
The quality of medicines in Pakistan is of major concern. DRAP is currently unable to effectively check the quality of all APIs and finished products through the available surveillance testing laboratories that are under governmental control. These materials are typically only tested to be quantified, and not for the identification of impurities, nor do they undergo any other pharmacopoeia tests. In addition, most of the laboratories involved in conducting these tests appear to contain out-dated instruments and materials, and there are concerns over levels of staff training and availability, as only a limited number of staff have been hired in recent years. This is despite such regulations being included in current laws [24].
The medicine registration process in any country is key to the availability of medicines that meet agreed quality targets. Concerns over the quality of generics produced in Pakistan is currently resulting in low exports [25] and impacting on patient care, especially for patients with chronic diseases.
There are also concerns that the basic and semi-basic industry involved in the manufacture of raw materials in Pakistan has not flourished due to unfavourable policies towards the protection or security of businesses. This is reflected by the fact that only 33 manufacturing units are currently involved in basic or semi-basic manufacturing of pharmaceuticals in Pakistan [26]. This compares with approximately 600 active licensed manufacturers of finished products in Pakistan [27]. In addition, among these 33, only seven manufacturing units currently appear active further demonstrating problems with current policies [26].
Objective
Considering these concerns, this study has assessed the quality of APIs in Pakistan and seeks to use the findings as a starting point for suggesting improvements in the registration process for oral generic tablets. We chose ibuprofen for our study in view of the extent to which it is prescribed in Pakistan, which is 12% by value of the analgesic market. The analgesic market currently has a growth of 20% per annum [28]. In addition, APIs of ibuprofen are produced both locally and imported. For ibuprofen, there are specific concerns relating to potential impurities in the light of pharmacopoeia specifications. The findings will subsequently serve as a guide to suggest improvements for the pharmaceutical drug registration process in Pakistan, to ensure good quality, safe, effective and affordable medicines are being produced that will help improve patient care in Pakistan, and potentially boost exports.
Methods
Collection of ibuprofen API samples
Twenty-seven samples of ibuprofen APIs used by manufacturers in Pakistan were obtained, together with their Certificate of Analysis (CoA). The CoA contained details regarding the results of tests and their values and limits (including assay values) of relevant batches, as well as information about the manufacturer. The US Pharmacopeia (USP) reference standard ibuprofen was obtained from Abbott Laboratories, Pakistan (originator manufacturer of ibuprofen). Coding was undertaken on all collected samples, with all samples stored in closed containers. Desiccators were used to avoid moisture absorption.
Quality assessment
The quality assessment of the ibuprofen samples was performed using the following methods:
1. Identification test
All samples were identified using the following two methods:
- FTIR (Fourier Transform Infrared) Spectroscopy. In this method, dry samples were used and placed in the instrument after cleaning with methanol. The HazMatID (Smiths Detection, USA) instrument was used for this purpose. The results from the HazMat (FTIR analyser) were interpreted from percentage resemblance, rather than being represented as concordant or not concordant with the reference spectrum.
- UV-Spectrophotometry. The UV identification test for ibuprofen is the ratio of the absorbance at 264 nm and 273 nm, reported again as percentage difference to the USP Reference Standard, and not as a pass/fail.
For all API samples and the USP Reference Standard (Abbott Laboratories, Pakistan), the solutions were prepared in 0.1 N Sodium Hydroxide with a concentration of 0.025 g per 100 mL, equivalent to 250 μg per mL of ibuprofen. Respective absorptivities at 264 nm and 273 nm on the anhydrous basis were noted.
For the UV-Spectrophotometer limits, the absorptivities were calculated on an anhydrous basis, and should not differ by more than 3.0% at 264 nm and 273 nm, as per USP limits.
The testing of ibuprofen against USP specifications was undertaken in one of Pakistan’s ‘Appellate Laboratory’, known to perform to the highest standards. The utilization of an USP approved laboratory to undertake the testing has significance in terms of the reliability of the results, negating the need to test the samples in other USP international laboratories in either Brazil or China or India.
2. Assay test
USP specifications mention the following limits for assay testing, ‘Ibuprofen (Active Pharmaceutical Ingredient) contains not less than 97.0 per cent and not more than 103.0 per cent of C13H18O2, calculated on the anhydrous basis’ [29]. The USP only documents one related or impurity substance, this is in contrary to the British Pharmacopoeia specifications where 18 substances are mentioned. However, the identification and characterization of related or impurity substances is out of the scope of this paper.
The Assay Test procedure
Mobile phase
Chloroacetic acid (4.0 g) was dissolved in water (400 mL) and then adjusted to a pH of 3.0 with ammonium hydroxide. Acetonitrile (600 mL) was added, then filtered, and degassed. Amendment or modifications were done as per the requirements of System Suitability.
Preparation of standard solution
A solution with concentration of about 12 mg per mL was prepared by dissolving USP Ibuprofen RS (accurately weighed) in Internal Standard Solution.
Assay preparation
1200 mg of ibuprofen, accurately weighed, was transferred to a 100 mL volumetric flask, diluted with Internal Standard Solution to volume, and mixed. Table 1 contains details of the chromatographic conditions.

The L1 column packing used was Octadecysilane C18 as C18 is generally more retentive than C8.
Test for system suitability
The peak response was recorded after repeated injections of five to six consecutive standard solutions before injecting the sample solutions. This was repeated after completion of work, to observe the consistency of performance of the system.
Acceptability criteria
The qualification criteria for the system suitability test was that there should be less than 2% Relative Standard Deviation (RSD) for five replicate injections of the standard solution, with not more than 2.5 tailing factors for the individual peaks.
Procedure
Equal volumes (approximately 5 μL) of the Standard preparation and the Assay preparation were separately injected into the chromatograph. Chromatograms were recorded and the response for the major peaks was measured.
The quantity of ibuprofen in mg was calculated using the formula: 100 C (RU/RS) where:
- C is the concentration, in mg per mL, of USP Ibuprofen RS in the Standard preparation; and
- RU is the peak response ratios obtained from the Assay preparation; and
- RS is the peak response ratios obtained from the Standard preparation.
Results
1. Identification test
Through FTIR (Fourier Transform Infrared) Spectroscopy:
Table 2 documents the percentage resemblance of the FTIR spectra of the 27 samples, to the USP standard spectrum. It also shows the percentage difference between the 27 samples and the USP standard spectrum, for the 264 nm and 273 nm peaks, recorded via UV-Spectrophotometry.
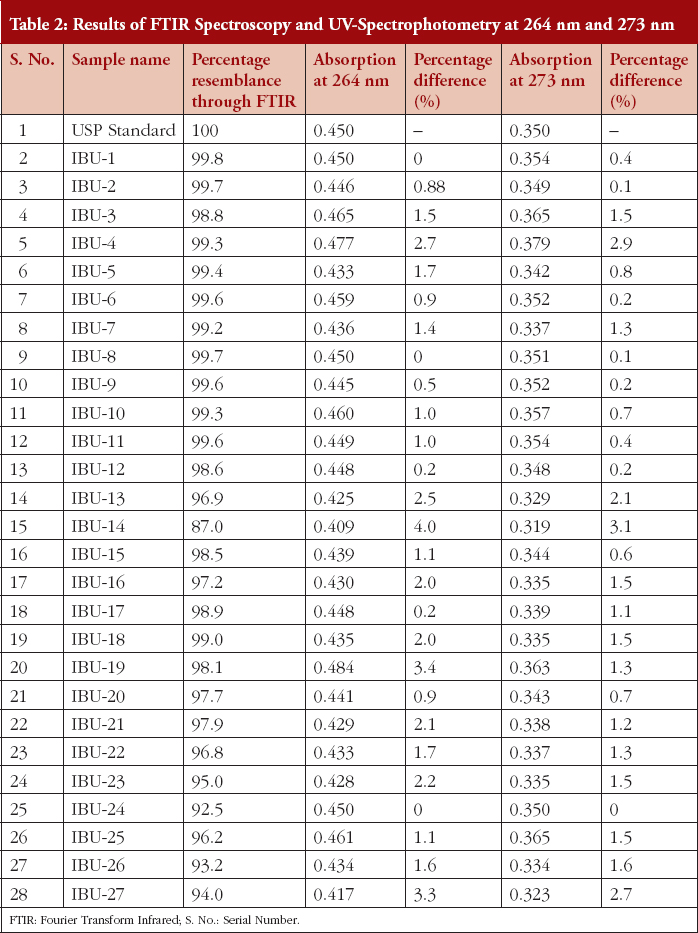
For measurements taken on the same day, precision ranged from 0.23% to 0.62%, and accuracy ranged from 99.6% to 100.3%. For measurements taken on different days, precision ranged from 0.24% to 0.52%.
2. Assay values
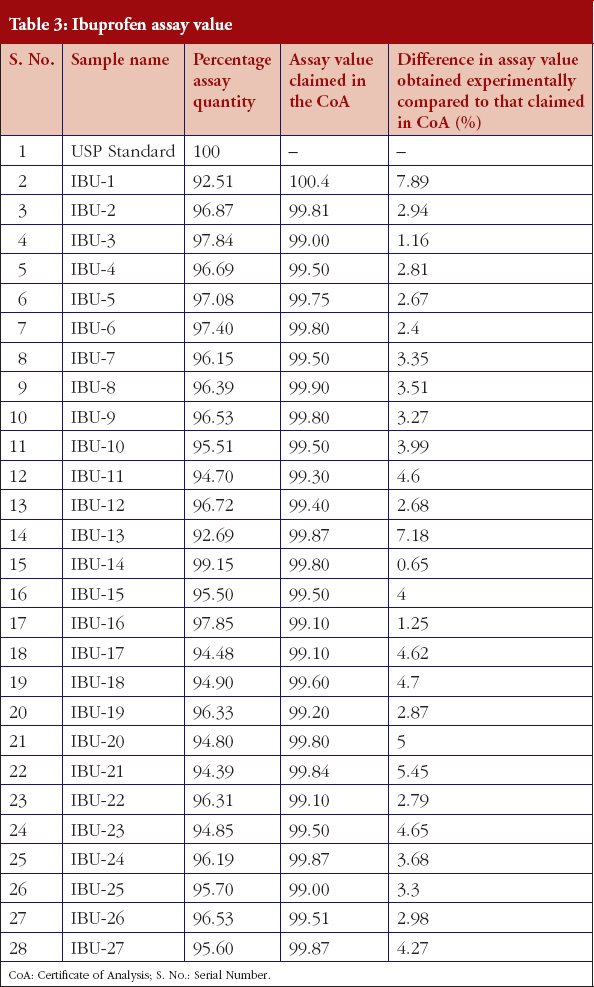
Table 3 documents the assay values of the 27 samples and the ibuprofen USP reference standard.
Discussion
The results reveal both positive and concerning aspects surrounding the APIs currently used in the production of generic ibuprofen tablets in Pakistan.
All samples, except sample IBU-14, passed the Identification test through FTIR and UV methods as per the requirements of USP specifications, see Table 2. Using the analytical methodology or assessment suggests that 96% API samples passed the test and can be approved and marketed as ibuprofen.
However, 22 out of 27 (81.5%) of the ibuprofen samples failed to comply with the USP assay limits, i.e. 97% to 103%. Interestingly, sample IBU-14 passed this test with highest percentage of assay value, i.e. 99.15%, see Table 3. IBU-14 also showed minimal difference in assay value to the values mentioned in the product CoA, see Table 3.
Secondly, one extra peak was noticed in the chromatograms of six (18.5%) of the samples, i.e. IBU-3, IBU-4, IBU-5, IBU-10, IBU-19 and IBU-25, at approximately the same time, i.e. between 3.1 to 3.3 minutes.
Thirdly, the comparison of assay values obtained using our methodology, versus those claimed by the manufacturer in their CoA, see Table 3, shows that none of the samples complied with the assay values claimed in their CoAs. Instead, three (11%) of the samples showed more than a 5% difference in assay value. In the majority of cases, the manufacturers of the finished products did not perform any testing on the API supplied. Instead, they typically rely on the CoA. This should be of concern to both regulators and manufacturers. Regulators in terms of the implementation of cGMP, while manufacturers should ethically and legally be responsible to follow cGMP for their finished products. In cases where product manufacturers perform tests on the APIs, when assay values of API are found to be lower than the prescribed limits, instead of rejecting the raw material or API, they typically use higher quantities in the production of ibuprofen tablets based on their own calculations, which raises concerns over the quality of products (Hussein S 2016, personal communication, November 8).
Our findings show a general failure of the current system of drug regulation in Pakistan that surrounds the quality of APIs used for drug production. This is in line with previous publications [5]. This will negatively impact on the quality of finished generic products for use in patients and will potentially compromise patient care. The results also show that quality assessment of multiple source medicines should not rely on assay testing alone. Other pharmacopoeia tests are also important, especially in cases where the optical activity and the presence of genotoxic, and other impurities, have a critical impact or role regarding the efficacy, safety and quality of medicines. Here, the failing sample of ibuprofen (IBU-14) passed the assay test with the highest value, while the sample failed the identification test. This invites scientific discussion regarding the value of current assay testing for generics in Pakistan, see Table 2. The results suggest that IBU-14 was not a pure API of ibuprofen. Instead, it may contain related substances or impurities which have a very close structural resemblance to the API of ibuprofen, and the HPLC method used could not identify these or discriminate them from the actual API of the drug [30]. This is why the USP does not mention the use of the HPLC method for the identification testing of ibuprofen API. Consequently, the USP compendium of methods for the identification of ibuprofen API, i.e. the FTIR and UV-Spectrophotometer methods, should be used in the future to assess the content of APIs in Pakistan.
The extra peak in the chromatograms, at 3.1 to 3.23 minutes, is also an important observation. When samples IBU-3, IBU-4, IBU-5, IBU-10, IBU-19 and IBU-25 (18.5%) were investigated for their source of manufacturing, it was found these samples were procured from only three sources. Two were in Pakistan (B and H) and one was in India (C). Further appraisal revealed that all APIs purchased from sources C and H showed this extra peak at the same time range. This illustrates the necessity to perform prequalification studies to evaluate the quality of the API from pharmaceutical manufacturers, before turning the raw materials into finished goods. However, only two out of seven (approximately 29%) API samples purchased from source B showed this extra peak. These results need to be further investigated for the characterization of this peak through evaluation of the route of synthesis or method of manufacturing of the API in order that potential corrective and preventive measures can be suggested for the future. It seems that this extra peak may be due to residual solvent or impurities remaining in the API. However, this needs further investigating before any definitive statements can be made. We are aware that both the British Pharmacopoeia and European Pharmacopoeia include a test for related substances for ibuprofen, with 18 potential impurities listed in the British Pharmacopoeia. However, as mentioned previously, the USP mentions only one specific impurity.
Assessment of the drug registration process and requirements in Pakistan
In view of the findings regarding the assessment of the current procedures concerning the registration of medicines in Pakistan, the technical requirements outlined in Box 1 are a potential way to improve the quality of medicines in Pakistan.
Currently, medicines being registered in Pakistan are not undergoing full evaluation of their safety and quality, especially in terms of their APIs [5]. Some of the suggested requirements, see Box 1, are not currently a mandatory part of the registration of medicines in Pakistan, e.g. the bioequivalence of generic medicines and even submission of the SmPC (Summary of Product Characteristics) and PIL (Patient Information Leaflet). Others, which are covered under the current rules and procedures, are also not being completely fulfilled, such as performing stability studies and validation studies. Ethically and legally, applicants should be bound to fulfill these commitments and thus perform these studies before marketing their medicines. However, in reality very few companies are complying with these commitments and performing such studies before marketing their medicines (S Hussein personal communication). There was a recent incidence of counterfeit medicines at the Punjab Institute of Cardiology which is thought to be a direct result of such deficiencies in the registration process and negligence in following cGMP [31, 32].
Despite these concerns, pharmaceutical manufacturers in Pakistan currently appear reluctant to perform additional tests or provide more comprehensive information about their medicines, during and after registration. This is thought to be due to the potential negative impact this could have on business (S Hussein, personal communication). In fact, the reverse may be true which would lead to improvement in the quality of generics for consumption in Pakistan, and a greater potential for export to other countries.
Recommendations
Here we outline a number of recommendations that should be considered by the Pharmaceutical Evaluation and Registration (PE & R) Division within DRAP, in consultation with Pakistani pharmaceutical companies, to improve the quality of generics produced by domestic manufacturers for use in Pakistan as well as for exportation.
These include a stepwise plan for the implementation of CTDs and new requirements in line with ICH standards, over one to three years, for example:
- Step 1: For new drugs, imported drugs and narcotic and psychotropic drugs. (Two to six months).
- Step 2: For biological (other than imported) and anticancer drugs. (Two to six months).
- Step 3: For anti-tuberculosis (TB), anti-human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDs) and antimalarial drugs (other than) and anticancer drugs. (Two to six months).
- Step 4: For all antibiotics. (Two to six months).
- Step 5: For all essential drugs (except over-the-counter [OTC]). (Two to six months).
- Step 6: For cardiovascular and anti-diabetes drug. (Two to six months).
- Step 7: For all remaining drugs. (Two to six months).
Alternatively:
- In the first phase, requirements already set out by the current rules and regulations should be implemented. This requires no additional effort by the manufacturers to carry out stability studies of API and Drug master files (open part) of the APIs and submission of the established SmPC and PIL of the product. This phase may take three to six months.
- In the second phase, there should be implementation of pertinent technical requirements which are not currently covered under the existing rules and regulations, such as bioequivalence studies. This will require more time as changes or improvements must be made to the infrastructure of DRAP.
Recently, the efforts of DRAP, in collaboration with USP and WHO, to develop the ‘Road Map for Strengthening the Registration System of Pharmaceutical Products and Biologicals for Human/Veterinary Use in Pakistan’, demonstrates a noticeable step towards improving the quality of medicines in Pakistan. We will be monitoring its progress and making additional recommendations if necessary.
Together with this, there is the ongoing process that seeks to convince pharmaceutical companies through discussions, seminars, and dialogue, of the need to adopt these new requirements for drug registration, to improve patient care and the potential for drug exports. The experiences of other countries that have started to accept such registration dossiers on CTD or e-CTD (Electronic Common Technical Document), should also be communicated within Pakistan to enhance acceptance of updated requirements. These countries include: Australia, Canada, China, Croatia, Japan, Saudi Arabia, Singapore, South Africa, Switzerland, the US, and all EU Member States [33]. In addition, comparison with the pharmaceutical manufacturing industry in Jordan whose population is only eight million compared with 201 million in Pakistan. Jordan currently has 16 pharmaceutical units compared with over 600 units in Pakistan [26, 34]. However, their exports are high with over 80% of produced medicines currently being exported over 60 countries [34, 35]. This includes more than US$4.5 billion alone to Saudi Arabia [35]. In contrast, Pakistani manufacturers in 2013 only exported US$1 billion due to concerns with poor quality [25, 36].
The implementation of ICH Standards should also be applicable to already registered medicines. These improvements may be achieved in the same stepwise manner as described above at the time of their application for renewal of registration starting over the coming year.
Other recommendations may include:
- Improved tracking of agreed implementation issues, maintained through dialogue sessions with pertinent pharmaceutical companies under political and governmental control, with the help of external organizations, such as WHO. This should help speed up the adoption of new processes to improve the quality of generics in Pakistan.
- During the transition period and prior to mandatory implementation, voluntary compliance with agreed new data requirements is advocated. Collection of BE (bioequivalence) data, should also be encouraged. This could be achieved through faster approval times and other beneficial regulatory aspects.
- Preparation of a minimum checklist, providing guidance to manufacturers on specific documentation requirements to reduce impurities and improve the quality of API raw materials. Internal staff within the regulatory agencies in Pakistan should familiarize themselves with, and gain an in-depth knowledge of the revised/new requirements. This can be done through attending internal and external training programmes and sharing information amongst peers.
- Providing manufacturers with additional motivation to hasten or oblige the adoption of these new requirements. This could include shorter approval times for pending applications/post-registration variations.
Manufacturers and DRAP may consider the following technical aspects whilst undergoing prequalification of API sources:
- Assessment of API dossier
- Inspection of manufacturing sites
- Random sampling and testing
- Details about handling of complaints and recalls
- Periodic or continuous checks to maintain the prequalification status
- Requirements for assessment of quality from DMF (Drug Master File) or APIMF (Active Pharmaceutical Ingredient Master File), together with the open part and closed part details.
If these measures are adopted, patients can expect good quality generics in the future. This is important, given the extent to which medicines in Pakistan are self-pay. Without such measures, patient care using generics and trust in the healthcare system will continue to be compromised, and there will be increased potential for adverse drug reactions [37]. Successful steps have already been taken to address concerns over counterfeit medicines in Pakistan, which need to continue [38]. The above considerations should be the next step to further improve the availability of safe, effective and affordable medicines in Pakistan. This is of great importance given the increasing prevalence of chronic illnesses and diseases in this country, where every third person over the age of 40 is vulnerable to a wide range of diseases [39], and 70% of the population lives on less than US$2 per day [40].
Conclusion
The results of this study document the concerns over the current regulations for assessment of the quality of drug raw materials (APIs) in Pakistan. These need to be urgently addressed to ensure good quality generics in Pakistan for patients, and to improve potential export opportunities. The adoption of WHO and ICH recommended CTD format and WHO prequalification guidelines, to improve the process of registration of medicines in Pakistan, should help improve the current system for registering medicines. Thus, this will enhance the availability of safe, effective and cost-effective generics for patients in Pakistan and other regions. This is starting to happen and will be monitored.
Competing interest: The authors declare that they have no conflicts of interest apart from those stated. No writing assistance was utilized in the production of this manuscript. The write-up of this paper was in part sponsored by the Karolinska Institutet.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Babar Khan1, BPharm, MPH, PhD
Brian Godman2,3, BSc, PhD
Ayesha Babar1, BPharm, MPhil
Shahzad Hussain4, BPharm, MPhil
Sidra Mahmood5, PharmD, MSc
Tahir Aqeel1, BPharm, MPhil
1Department of Pharmacy, University of Lahore, Jinnah Avenue, Islamabad Campus, Pakistan
2Department of Laboratory Medicine, Division of Clinical Pharmacology, Karolinska Institute, Karolinska University Hospital Huddinge, SE-14186 Stockholm, Sweden
3Strathclyde Institute of Pharmacy and Biomedical Sciences, Strathclyde University, Glasgow G4 ORE, UK
4National Institute of Health, House No. C13, NIH Colony, Chak Shahzad, Park Road, Islamabad, Pakistan
5Scotman Pharmaceuticals Pvt Ltd, 1-10/3, Industrial Area, Islamabad – 4400, Pakistan
References
1. Chowdhry NJ. Growth challenges of pharmaceutical industry in Pakistan. PPMA. Available from: http://health-asia.com/conferences/2014/Presentations/Day1/PharmaConvention/02-Nasir-javaid.pdf
2. European Commission (EC) Trade-related Technical Assistance Programme (TRTA) for Pakistan [homepage on the Internet]. [cited 2016 Nov 8]. Available from: http://www.tradecapacitypakistan.com/new/pdf/itc/SS2.pdf
3. Hafeez-Ur-Rehman. Manual of drug laws 2010. 2nd ed. Karachi: Pioneer Book House; 2010. 216-253.
4. Manual of Drug Laws. The Drug Act, 1976 [homepage on the Internet]. [cited 2016 Nov 8]. Available from: http://medisure.com.pk/drug_law/DrugAct+Rules/index.htm
5. Zaidi S, Bigdeli M, Aleem N, Rashidian A. Access to essential medicines in Pakistan: policy and health systems research concerns. PloS One. 2013;8(5):e63515.
6. World Health Organization. WHO good manufacturing practices for pharmaceutical products: main principles. Annex 2 [homepage on the Internet]. [cited 2016 Nov 8]. Available from: http://www.who.int/medicines/areas/quality_safety/quality_assurance/TRS986annex2.pdf
7. Jamshed SQ, Hassali MA, Ibrahim MI, Babar ZU. Knowledge attitude and perception of dispensing doctors regarding generic medicines in Karachi, Pakistan: a qualitative study. J Pak Med Assoc. 2011;61(1):80-3.
8. Jamshed SQ, Ibrahim MI, Hassali MA, Masood I, Low BY, Shafie AA, et al. Perception and attitude of general practitioners regarding generic medicines in Karachi, Pakistan: a questionnaire based study. South Med Rev. 2012;5(1):22-30.
9. Riaz H, Godman B, Hussain S, Malik F, Mahmood S, Shami A, Bashir S. Prescribing of bisphosphonates and antibiotics in Pakistan: challenges and opportunities for the future. J Pharm Health Serv Res. 2015;6:111-21.
10. Shrank WH, Hoang T, Ettner SL, Glassman PA, Nair K, DeLapp D, et al. The implications of choice: prescribing generic or preferred pharmaceuticals improves medication adherence for chronic conditions. Arch Intern Med. 2006;166(3):332-7.
11. Corrao G, Soranna D, La Vecchia C, Catapano A, Agabiti-Rosei E, Gensini G, et al. Medication persistence and the use of generic and brand-name blood pressure-lowering agents. J Hypertens. 2014;32(5):1146-53.
12. Simoens S, Sinnaeve PR. Patient co-payment and adherence to statins: a review and case studies. Cardiovasc Drugs Ther. 2014;28(1):99-109.
13. Kesselheim AS, Misono AS, Lee JL, Stedman MR, Brookhart MA, Choudhry NK, et al. Clinical equivalence of generic and brand-name drugs used in cardiovascular disease: a systematic review and meta-analysis. JAMA. 2008;300(21):2514-26.
14. Gagne JJ, Choudhry NK, Kesselheim AS, Polinski JM, Hutchins D, Matlin OS, et al. Comparative effectiveness of generic and brand-name statins on patient outcomes: a cohort study. Ann Internal Med. 2014;161(6):400-7.
15. Paton C. Generic clozapine: outcomes after switching formulations. Br J Psychiatry. 2006;189(2):184–5.
16. Veronin MA. Should we have concerns with generic versus brand antimicrobial drugs? A review of issues. J Pharm Health Serv Res. 2011;2(3):135-50.
17. Godman B, Wilcock M, Martin A, Bryson S, Baumgärtel C, Bochenek T, et al. Generic pregabalin; current situation and implications for health authorities, generics and biosimilars manufacturers in the future. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(3):125-35. doi:10.5639/gabij.2015.0403.028
18. Corrao G, Soranna D, Arfe A, Casula M, Tragni E, Merlino L, et al. Are generic and brand-name statins clinically equivalent? Evidence from a real data-base. Eur J Intern Med. 2014;25(8):745-50.
19. Baumgärtel C, Godman B, Malmström R, Andersen M, et al. What lessons can be learned from the launch of generic clopidogrel? Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(2):58-68. doi:10.5639/gabij.2012.0102.016
20. Godman B, Baumgärtel C. Are generic immunosuppressants safe and effective? BMJ. 2015;350:h3248.
21. The Gazette of Pakistan. 13 November 2012 [homepage on the Internet]. [cited 2016 Nov 8]. Available from: www.na.gov.pk/uploads/documents/1352964021_588.pdf
22. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). ICH guidelines [homepage on the Internet]. [cited 2016 Nov 8]. Available from: http://www.ich.org/products/guidelines.html
23. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). CDT. M4 : The common technical document [homepage on the Internet]. [cited 2016 Nov 8]. Available from: http://www.ich.org/products/ctd.html
24. Ramesh T, Saravanan D, Khullar P. Regulatory perspective for entering global pharma markets. Pharma Times. 2011;43(9):15-20.
25. Ikram J. Manufacturers say export of medicines declining. DAWN. 4 July 2015.
26. Drug Regulatory Authority of Pakistan. Updated list of pharmaceuticals manufacturers [homepage on the Internet]. [cited 2016 Nov 8]. Available from: www.dra.gov.pk/
27. Aamir M, Khalid Zaman. Review of Pakistan pharmaceutical industry: SWOT analysis. Int J Bus Inform Tech. 2011;1(1):114-7.
28. Analgesics in Pakistan. Euromonitor International. Sep 2016.
29. U.S. Pharmacopeial Convention. USP Ibuprofen updated list of pharmaceuticals manufacturers [homepage on the Internet]. [cited 2016 Nov 8]. Available from: http://www.usp.org/sites/default/files/usp_pdf/EN/USPNF/errata467Ibuprofen.pdf
30. Zolner W. Critical aspects of HPLC. Eagle Analytical Services. November 2014.
31. 2012 Pakistan fake medicine crisis. Medicines. 25 July 2012.
32. Rias A. Pharmaceuticals: drug or poison. theHealth. 2012;3(2):36-8.
33. Implementation status of CTD, eCTD, and paper-free submissions: a global overview. Thomson Reuters.
34. The Jordanian Association of Pharmaceutical Manufacturers [homepage on the Internet]. [cited 2016 Nov 8]. Available from: http://www.japm.com/Public/English.aspx?Lang=2&Page_Id=194&Menu_ID=6&Menu_Parent_ID=-1&type=R
35. Jordan Economic and Commerce Bureau [homepage on the Internet]. [cited 2016 Nov 8]. Available from: http://www.jordanecb.org/Public/English.aspx?Site_Id=1&Page_Id=551&menu_id=38
36. Indian Council for Research on International Economic Relations. Ahmed V, Batool S. India-Pakistan trade: a case study of the pharmaceutical sector. Working paper 291 [homepage on the Internet]. [cited 2016 Nov 8]. Available from: http://www.slideshare.net/vahmed/indiapakistan-trade-a-case-study-of-the-pharmaceutical-sector
37. Mahmood KT, Amin F, Tahir M, Haq IU. A need for best patient care in Pakistan. A review. J Pharm Sci & Res. 2011;3(11);1566-84.
38. Stopping fake drugs from Pakistan is too late for victims. Bloomberg.
39. Riaz H, Godman B, Bashir S, Hussain S, Mahmood S, Malik F, et al. Evaluation of drug use indicators for non-communicable diseases in Pakistan. Acta Pol Pharm. 2016;73(3):787-94.
40. Shaikh BT, Ejaz I, Mazhar A, Hafeez A. Resource allocation in Pakistan’s health sector: a critical appraisal and a path toward the Millennium Development Goals. World Health Popul. 2013;14(3):22-31.
|
Author for correspondence: Brian Godman, BSc, PhD, Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow G4 0RE, UK; Division of Clinical Pharmacology, Karolinska Institute, Karolinska University Hospital Huddinge, SE-14186 Stockholm Sweden |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2016 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



This article sheds light on the quality concerns with the pharmaceutical, like API in alfa chemistry. With the majority of raw materials being imported from other countries and the lack of updates to GMP requirements, there are concerns over the safety and efficacy of medicines manufactured in the country. The inconsistency between the information required for medicine authorization and the lack of implementation of internationally harmonized standards also highlight the weaknesses in the registration process. In addition, the shortage of testing laboratories and outdated instruments further exacerbates these issues. These concerns have resulted in low exports and impacted patient care, particularly for those with chronic diseases. It is crucial for the Pakistani government to address these concerns and prioritize the implementation of updated regulations to ensure the availability of quality medicines for their citizens.
Dear Mindy Hausler,
We very much appreciate your kind feedback. We have informed the authors of your comments.
Thank you for your interest in GaBI. Please enjoy the high-quality information and content published by GaBI (GaBI Online and GaBI Journal).
GaBI Journal Editorial Office