Biosimilars markets: US and EU compared
Published on 2020/05/26
Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):90-2.
|
Abstract: |
Submitted:29 February 2020; Revised: 29 April 2020; Accepted: 4 May 2020; Published online first: 18 May 2020
The US and Europe have a number of key differences in the way in which they approve biosimilar drugs. These were discussed at a recent conference [1], in addition to some strategies that can be used to address healthcare provider and patient concerns regarding biosimilars.
US and EU biosimilars markets compared
Comparisons of the time taken to authorize (or recommend approval of) a selection of nine biosimilars by the European Medicines Agency (EMA) and the US Food and Drugs Administration (FDA) are shown in Table 1.

EMA approved their first biosimilar, somatropin, in April 2006. The following year, an epoetin alfa biosimilar was approved by EMA in August, followed by FDA approval in May 2018; 11 years later. Similarly, a filgrastim biosimilar was approved first in the European Union (EU) in September 2008, followed by US approval in March 2015. Filgrastim was the first biosimilar approved in the US, nine years after EMA’s first approved biosimilar, somatropin.
However, more recently, approval times in the US have caught up. For example, a bevacizumab biosimilar was first approved in the US in September 2017, while EMA approved a bevacizumab biosimilar a few months later in January 2018.
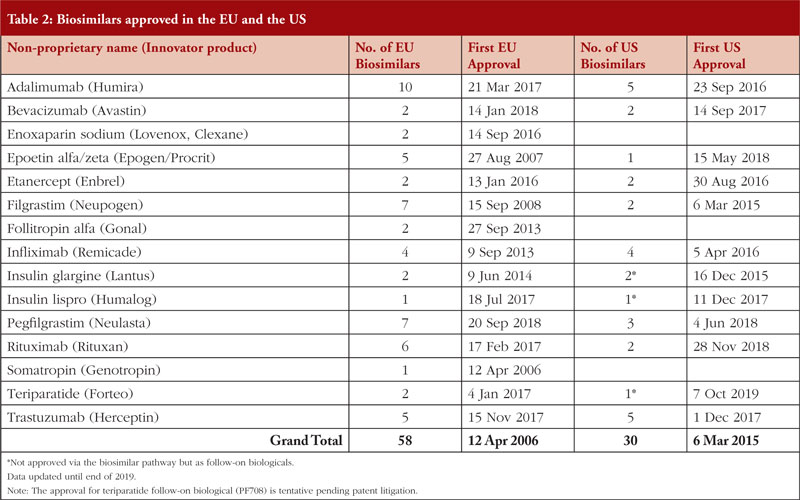
Table 1, however, does not include five biologicals shown in Table 2, i.e. enoxaparin sodium, follitropin alfa, insulin glargine, insulin lispro, somatropin, which were not approved via the biosimilar pathway in the US [2] at the time of EU approval. By end of 2019, there are 26 biosimilars approved in the US and 58 approved in the EU [3].
In Europe, biosimilar competition has significantly reduced prices for biologicals, as demonstrated by the change in price (per treatment day) in the EU in the year of the first biosimilar launch, see Figure 1.

Total market prices for erythropoietin, for example, decreased by around 30%, which was similar to the reductions in prices seen for granulocyte colony-stimulating factor (G-CSF) after the launch of a biosimilar version.
In the US, the opportunities for biosimilars can be particularly lucrative. For an overview of biosimilar opportunities in the US until 2023, see Figure 2.

There are 71 patents on biologicals due to expire in the US by 2023, with a total of US$55 billion in sales open to competition from biosimilars manufacturers [1].
Together, expiry of the patents on Humira (adalimumab), Kadcyla (trastuzumab), Lumizyme (alglucosidase alfa) and Stelara (ustekinumab) are worth an estimated US$20 billion to biosimilars manufacturers.
Patents on Amevive (alfacept), Lemtrada (alemtuzumab) and Rituxan (rituximab), meanwhile, have already expired in the US. Biosimilars for these products have an estimated value of US$4.5 billion. Of these, so far only rituximab (Pfizer and Celltrion/Teva) and trastuzumab (Celltrion/Teva; Amgen; Biocon/Mylan; Merck/Samsung Bioepis; Pfizer) have licensed biosimilars in the US [2]. Amgen/Allergan have additionally submitted an application to FDA for their rituximab biosimilar [4].
Addressing concerns about biosimilars
Although the biosimilars market is advancing well in both EU and the US, concerns about the use of biosimilars remain widespread. A number of strategies can be employed to address the concerns of both healthcare providers and patients.
Utilizing the EU’s experience in biosimilars can be helpful, especially in cases where there are limited data. Almost two times as many biosimilars have been approved in the EU compared to the US, see Table 2.
The EU monitoring system for adverse events, in particular, can help to provide evidence for the safety of biosimilars. Furthermore, EMA states that evidence it has acquired over 10 years shows that biosimilars can be used ‘as safely and effectively in their approved indications as their reference biological medicines’ [5]. The European experience is testament to the value of multi-stakeholder participation and engagement at every stage of the process, from the regulatory framework through education all the way to procurement policy decisions and treatment guidelines [6].
Gaining support from physician leaders within an organization (or other key opinion leaders) can also help to alleviate concerns about biosimilars, while educational initiatives aimed at healthcare providers and patients can help to minimize the nocebo effect (when negative expectations of a treatment, in this case a biosimilar, lead the patient to believe it is less effective).
Training healthcare providers to be well informed and confident in the use of biosimilars can lead to the transfer of confidence to the patient. FDA materials on biosimilars can be useful for such educational purposes.
A further recommendation is to transition patients to biosimilars in a conservative manner in order to gain experience and support. Monitoring the outcomes of conversion can also improve confidence in the use of biosimilars. Finally, it is important to advocate for comparable patient assistance programmes with biosimilar manufacturers.
Competing interests: None.
Provenance and peer review: Commissioned; internally peer reviewed.
Eleanor Bird, MSc; GaBI Journal Editor; Michelle Derbyshire, PhD, GaBI Online Editor
References
1. Gutierrez A. Payor strategies to drive biosimilars access and savings. GRx+Biosims Conference 2019; 4 6 November 2019; North Bethesda, Maryland, USA.
2. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in the US. [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 29]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-the-US
3. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe. [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 May 9]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
4. GaBI Online – Generics and Biosimilars Initiative. Rituximab biosimilar ABP 798 submitted to FDA [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 29]. Available from: www.gabionline.net/Biosimilars/News/Rituximab-biosimilar-ABP-798-submitted-to-FDA
5. GaBI Online – Generics and Biosimilars Initiative. Improving understanding of biotherapeutics and biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2020 Apr 29]. Available from: www.gabionline.net/Biosimilars/General/Improving-understanding-of-biotherapeutics-and-biosimilars
6. Reilly MS, Schneider PJ. Policy recommendations for a sustainable biosimilars market: lessons from Europe. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):76-83. doi:10.5639/gabij.2020.013
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2020 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


