Clinical development, immunogenicity, and interchangeability of follow-on complex drugs
Published on 2014/01/22
Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):71-8.
|
Abstract: |
Submitted: 16 January 2014; Revised: 24 February 2014; Accepted: 24 February 2014; Published online first: 10 March 2014
Introduction
The Biologics Price Competition and Innovation (BPCI) Act of 2009 was instituted to create an abbreviated pathway for approval of biosimilar drugs [1]. In 2014, the biosimilar pathway is still evolving; at this writing, the US Food and Drug Administration (FDA) has issued three draft guidelines for manufacturers seeking approval of biosimilar drugs [2–4]. Regulatory authorities agree that pre-approval evaluation of biosimilar drugs must be held to a higher standard than generic versions of small-molecule drugs because of their complexity and immunogenicity [5–7]. Non-biological complex drugs (NBCDs) have the molecular complexity of biological drugs, are immunogenic, and developing follow-on NBCDs poses many of the same scientific challenges associated with biosimilar drugs [8–10]. NBCDs typically contain heterogenous mixtures of closely related, macromolecular, nanoparticulate components that cannot be isolated, quantified, and/or entirely characterized physicochemically using available analytical technology [11]. As is true for biological drugs, consistent NBCD activity and quality typically rely on strictly controlled manufacturing procedures [12–14], such that even small differences in the manufacture of a follow-on NBCD from that of the originator product can increase the risk of safety problems or reduced therapeutic efficacy [13, 15, 16].
Currently, follow-on NBCDs can be approved under the generics pathway established for traditional small-molecule drugs via an abbreviated new drug application (ANDA [505(j) application]), or under section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act (FFDCA) [17]. Acknowledging that this pathway may not address the scientific challenges of ensuring the safety and efficacy of follow-on NBCDs [11, 18], FDA has made ‘equivalence of complex drugs’ a Generic Drug User Fee Amendment (GDUFA) Regulatory Science Priority Initiative for fiscal year 2014 [19].
A key aspect of pending legislation for biosimilars and follow-on NBCDs will be the development of science-based policies for interchangeability and drug substitution. The BPCI Act makes clear that biosimilarity does not imply interchangeability or substitutability [1]. Unlike generic copies of small-molecule drugs, biosimilars and follow-on NBCDs will not be identical to the innovator products. Because of their complexity and because the manufacturing method used to produce the innovator drug is often proprietary, some variation in alternate-sourced products is inevitable. Slight but clinically meaningful differences between originator and follow-on NBCDs may make interchangeability unfeasible. By law, to gain approval for interchangeability for biological drugs, the risk in terms of safety or diminished efficacy of alternating or switching between the generic product and the reference product must be no greater than continuing to use the reference product [1].
Scientific issues related to therapeutic equivalence of NBCDs
The ANDA generic drug pathway for small-molecule drugs requires proof of therapeutic equivalence of the generic to the innovator product, i.e. pharmaceutical equivalence (identical active substances, dosage form, strength, route of administration, labelling, quality, performance characteristics, and intended use), and bioequivalence (comparable pharmacokinetics in healthy humans) [17]. For some NBCDs, full proof of pharmaceutical equivalence is impossible, since two drugs cannot be shown to have identical active substances if the active substance has not been identified and the mechanism of action of the reference drug remains unknown [8, 10, 13]. Gross characterization of drug composition showing similarities in certain vectors, e.g. molar ratio or molecular weight distribution of constituents, does not guarantee similarity of other product characteristics [13, 20]. Similarly, bioequivalence cannot be established for many NBCDs because their biological activity is not correlated to serum pharmacokinetics [10, 21, 22]. These drugs typically comprise nanoparticle-size substructures that release or form the active ingredient, which is then transported to the targeted tissue.
Additionally, follow-on NBCDs cannot be presumed to have the same immunogenic profiles as innovator complex drugs [23]. The ability to predict drug-induced immunogenicity of uncharacterized NBCDs is limited, because immunogenicity is subject to influence by many variables. Patient-related factors such as genetic background, immune status, and the disease under treatment will influence the immunogenic response to treatment [24]. Autoimmune diseases can augment the immune response to immunogenic drugs. Product- and manufacturing-related factors also influence immunogenicity [5, 25, 26]; minor but key changes to the synthesis or manufacture of follow-on protein- and peptide-based NBCDs can lead to formation of aggregates or other impurities that can enhance drug-related immunogenicity and be immunogenic in their own right [26, 27].
Two products purported to be the same drug can produce antibodies with varying specificity such that one drug produces neutralizing antibodies (NABs) and the other does not [14]. When switching between a follow-on drug and the reference product (or among follow-on products), pre-existing antibodies to one NBCD could neutralize the efficacy of an analogous product. NABs that decrease drug efficacy can develop months or years after beginning treatment [28, 29]. For example, clinically important NABs associated with interferon-beta (IFNβ) therapy for MS generally develop after 12 to 18 months of treatment [30], and the clinical effects of decreased efficacy may take years to detect, resulting in irreversible disability progression that might have been avoided by performing regular antibody assessments [31].
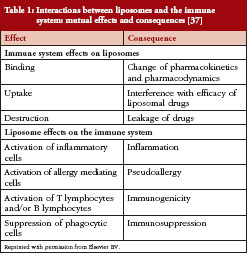
Liposomes, iron-carbohydrate complexes, and glatiramoids
The 2014 GDUFA initiative regarding equivalence of complex drugs specifically mentions generic versions of (among others) liposomal drug formulations, e.g. Doxil (doxorubicin HCl liposome); iron-carbohydrate complexes, e.g. Venofer (iron sucrose); and products that contain complex peptide mixtures and peptides, e.g. Copaxone (glatiramer acetate) [19]. These NBCD classes exemplify challenges to the classic abbreviated pathway for generic drug approval and indicate a need for increased pre-approval assessment for follow-on NBCDs.
Liposomal drug formulations
Liposomal drug formulations act as carrier vehicles to deliver active agents to a specific body site. Nanoparticles of the bioactive agent encapsulated in vesicles composed of a phospholipid bilayer act as targeted antigen delivery systems to induce therapeutic humoral and cell-mediated immune responses [22]. As vaccines, synthetic antigenic peptides in liposomal formulation induce autoantibodies for prophylaxis of chronic conditions, such as hypertension [32]. Liposomal formulations of anticancer drugs allow antibody- or ligand-mediated targeting specifically to tumour cells, to increase therapeutic effects while reducing toxicity [33].
The physicochemical properties of liposomal vaccines – method of antigen attachment, lipid composition, bilayer fluidity, particle charge, and other properties – strongly influence the immune responses to them [22, 34], see Table 1. Thus, small differences in particles or complex attributes in follow-on versions of liposomal drugs could alter the activity of the drug, its distribution profile, and/or its persistence at tissue/cellular or subcellular levels to a clinically meaningful extent [35]. Currently, little is known about the cellular distribution of lipid-modified
peptides [22].
Consistent plasma concentrations of the active substance in two liposomal formulations does not guarantee similar efficacy or safety of the two products, since nanoparticles of active drug may distribute differently in tissues and cells [13, 35]. To investigate whether a conventional bioequivalence approach could ensure therapeutic equivalence of liposomal products, the pharmacokinetics, efficacy, and toxicity of six formulation variants of the originator PEGylated liposomal doxorubicin product (Doxil/Caelyx, Janssen-Cilag Pty Ltd) were prepared differing in composition and liposome size and evaluated in preclinical models for antitumour activity and toxicity [36]. Although some formulations demonstrated similar plasma pharmacokinetics and systemic exposure of doxorubicin, they exhibited different antitumour activity and toxicity profiles. Investigators concluded that a conventional bioequivalence approach is not appropriate for establishing therapeutic equivalence of a generic product.
Augmenting immunogenicity is key to the therapeutic activity of many liposomal preparations. However, some therapeutic liposomes are recognized by the immune system as foreign, likely because the phospholipid vesicles of the liposome mimic the size and shape of pathogenic microbes [37], leading to a variety of adverse immune reactions. Hypersensitivity reactions to liposomal drugs appear to be primarily mediated through complement activation triggered by an immune reaction to liposome surface charge or topography [37].
Detecting clinically meaningful differences in the therapeutic activity, toxicity, and immunogenicity of a follow-on liposomal drug may require nonclinical and clinical studies. A reflection paper issued by the European Medicines Agency (EMA) on data requirements for follow-on versions of liposomal products indicates clinical data for these products will be considered on a case-by-case basis [38]. Currently, EMA has not approved any follow-on versions of liposomal drugs.
Iron-carbohydrate drugs
Intravenous (IV) iron products are used to treat iron deficiency anaemia in patients undergoing chronic haemodialysis and receiving supplemental EPO therapy and in people with iron-deficiency anaemia associated with chronic blood loss or impaired iron absorption. The chemical structures of parenteral iron agents have not been characterized in full detail. Venofer (iron sucrose, Vifor Inc) and Ferrlecit (iron gluconate, Sanofi) comprise nanoparticle-sized iron cores surrounded by a complex carbohydrate layer. Because the physicochemical and biological properties of iron-carbohydrate compounds depend on their manufacturing processes, subtle structural modifications during manufacture may affect drug stability; if weakly bound iron dissociates prematurely it can catalyse the generation of reactive oxygen species leading to oxidative stress and inflammation [39]. Moreover, any variation in mean/median size and size distribution of the iron-carbohydrate nanoparticles can result in a generic product with different physicochemical properties and different biopharmaceutical profile with respect to pharmacokinetics and biodistribution compared with the originator drug [21]. In fact, animal studies show differences between the originator iron sucrose product (Venofer) and iron sucrose similar (ISS) products with increased markers of inflammation and increased serum iron and transferrin saturation levels in animals receiving the ISS [13, 40].
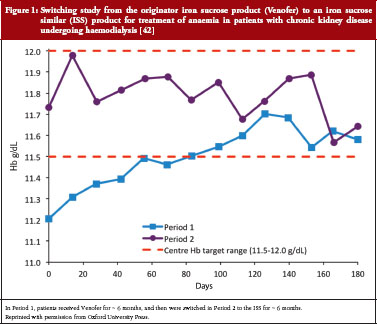
Despite these differences and the inability to completely characterize these drugs, and with no nonclinical or clinical studies to establish their therapeutic equivalence to the innovator drug, ISS products gained marketing approval via the small-molecule drug generic pathway [41]. Subsequently, in controlled trials in anaemic patients undergoing haemodialysis, ISS use was associated with reduced efficacy and the potential for increased safety risk related to iron overload [42, 43]. Clinically meaningful differences have been demonstrated when patients were switched to an ISS from Venofer. A switching study in which 75 stable haemodialysis patients taking Venofer for at least six months switched to an ISS product for six months resulted in decreased haemoglobin levels and reduced iron indices despite higher doses of the ISS [42], see Figure 1.
Iron-carbohydrate products can cause life-threatening or fatal hypersensitivity reactions, especially in pregnant women [44, 45]. The immunologic basis of allergic hypersensitivity to iron agents remains unknown [44]. Substitution of Venofer with an ISS at the pharmacy level (without physician or patient knowledge) was associated with hypersensitivity reactions and hospitalization in subjects who previously tolerated the originator drug [41]. Safety concerns surrounding all IV iron products led to recommendations of stronger measures to manage and minimize the risk of hypersensitivity [45]. The recommendations state that every dose of IV iron administered should be monitored for potential hypersensitivity reactions, even if previous administrations were well tolerated.
Both FDA and EMA have indicated that follow-on versions of iron sucrose (FDA) and nanoparticulate iron medicinal products (EMA) are not suitable for approval through the classic generic approval pathway [21, 46]. Neither agency has indicated what clinical evaluation will be required for approval of follow-on products.
Glatiramoids
The prototype glatiramoid, Copaxone, (Teva Pharmaceutical Industry) is a complex heterogenous mixture of synthetic proteins and polypeptide nanoparticles with immunomodulatory activity approved for treatment of relapsing-remitting multiple sclerosis (RRMS) [10, 47–50]. The active ingredient in Copaxone, glatiramer acetate, comprises a potentially incalculable number of unidentified active peptide moieties that are not characterizable with available technology [10], although the amino acid sequences in Copaxone are not entirely random [8]). The mechanism of action of Copaxone is not fully elucidated but the drug is thought to act as an antigen-based therapeutic vaccine [51–53]. Pharmacokinetic data are uninformative for glatiramoids because the polypeptides in a glatiramoid mixture are hydrolysed at the drug injection site into unidentifiable peptide fragments that stimulate proliferation of glatiramoid-specific immune cells, which migrate to the central nervous system where they ameliorate auto-immune destruction of myelin [54, 55]. Therefore, blood levels of the glatiramoid or its hydrolysis products are not indicative of drug activity.
Glatiramoids appears to act as altered peptide ligands (APL) of encephalitogenic epitopes within myelin basic protein (MBP), an autoantigen implicated in MS [56]. Decades of clinical use demonstrate that Copaxone does not contain encephalitogenic epitopes and does not induce auto-reactive antibodies [48]. However, the same cannot be assumed for a follow-on glatiramer acetate product. In the last two decades, other APLs of MBP epitopes have been studied for use as therapeutic vaccines in MS. Clinical development of at least two APLs of MBP antigenic peptides was halted due to adverse events indicative of auto-reactive antibodies (i.e. immediate-type hypersensitivity reactions [57]) or substantial expansion of pro-inflammatory T cells that were cross-reactive with the MBP autoantigens [58].
Because Copaxone works as a therapeutic vaccine, anti-drug antibodies are detectable in all treated patients [48, 49, 52]. These antibodies, however, do not neutralize biological activity or clinical efficacy and are not associated with local or systemic adverse effects in RRMS patients receiving chronic treatment [49]. Anti-Copaxone antibody titers and isotypes change over time with repeated drug administration [48, 49, 59]. Although anti-Copaxone antibodies are predominantly of the IgG subclasses over time [48, 49, 60, 61], there have been rare reports of anti-Copaxone IgE antibodies associated with anaphylactic reactions that can arise up to 10 months to a year after treatment initiation, with no symptomology beforehand to signal hypersensitivity [62, 63].
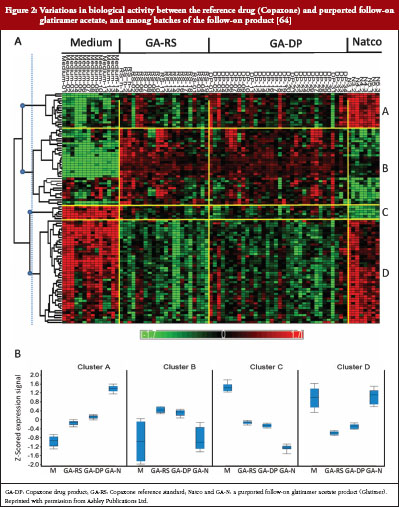
A purported follow-on product of glatiramer acetate is currently marketed in India and the Ukraine (Glatimer, Natco Pharma Ltd, Hyderabad, India). There are no published data of the safety, efficacy, or immunogenicity of this product at this writing. In analytical tests, this product demonstrated physicochemical differences from Copaxone and poor batch-to-batch reproducibility [8, 20, 64]. When activated ex vivo with Glatimer, splenocytes from GA-treated mice showed distinctly different gene transcription profiles among different batches, and between Glatimer and Copaxone, see Figure 2 [64].
A comparability trial of a generic glatiramer acetate product (GTR, Synthon VB) and Copaxone is currently underway in RRMS patients [65]. The GATE trial (ClinicalTrials.gov NCT01489254) is a 24-month study comprising a 9-month, placebo-controlled, active-comparator phase followed by a 15-month open-label phase in which all participants remaining in the study receive GTR. Characterizing the immunogenicity of GTR is not an objective of the study because the protocol suggests that anti-GTR antibodies will be the same as anti-Copaxone antibodies; specifically, that because anti-Copaxone antibodies are not neutralizing, anti-GTR antibodies will not be neutralizing either [65]. This assumption requires verification: GTR may be shown to be a close approximation to Copaxone at best and small differences in amino acid sequences or of protein folding in GTR could generate an antibody repertoire with different, isotopes, specificities, and affinities from those of anti-Copaxone antibodies, with variable consequences on patient safety and response to therapy [10, 13, 66, 67]. Accordingly, experts in the field of MS agree that the immunogenicity of follow-on versions of Copaxone cannot be assumed and should be established for each formulation [23].
According to the study protocol, the assessment of the immunogenicity of GTR is to compare proportions of subjects who develop anti-drug antibodies after receiving GTR or Copaxone [65]. As antigen-based therapeutic vaccines, antibody development to either drug would be expected in 100% of treated participants. Thus, it will not be possible to compare efficacy outcomes in patients free from anti-GTR antibodies with outcomes in GTR-antibody-positive patients to determine potential formation of NABs.
In the US, at least three manufacturers have filed ANDAs for follow-on glatiramer acetate products with FDA under the small-molecule generic pathway [68–71]. The manufacturers maintain that these products will be interchangeable with Copaxone, despite the fact that the first clinical exposure to these products in MS patients will occur post-approval. Given the complexity, unknown mechanism of action, uncharacterized epitopes, and strong immunogenicity of Copaxone; the variable nature of RRMS disease activity; and the inter- and intra-patient variability of antibody responses to immunogenic drugs; adequate testing of the immunogenicity of uncharacterized follow-on glatiramoid products in MS patients should precede approval and marketing of these products.
Considerations for approval of follow-on NBCDs
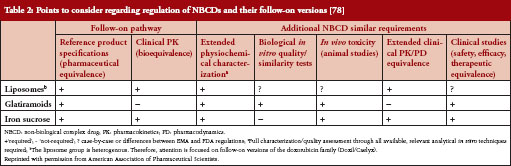
The generic approach should be limited to products that can be fully characterized and allow prediction of biological effects with pharmacokinetics data as surrogates for clinical efficacy [11]. Because NBCDs have many of the same features as biologicals, it seems prudent to extend guidelines for biosimilar products to follow-on NBCDs [2, 5, 24, 72]. When it is not possible to prove bioequivalence of follow-on NBCDs, requiring non-clinical and clinical testing can ensure therapeutic equivalence between NBCDs and the reference drug. Comparability evaluations for a follow-on NBCD should include physicochemical properties, impurities, biological activity, pharmacokinetics, efficacy, and safety, see Table 2. The extent of testing needed to establish adequate similarity between an originator drug and a follow-on product will likely depend on NBCD complexity, mode of action (if known), and the potential for toxicity. The risks of free substitution between an uncharacterized, immunogenic NBCD and a follow-on product will remain unknown without a clinical crossover study that provides direct evidence that repeated switching between the reference and the generic drug has no negative impact.
For immunogenic NBCDs, it may be necessary to ensure that the immunologic and immunogenic safety of the follow-on NBCD is comparable to that of the reference drug in clinical studies in patients with the disease under study. Considerable inter-individual variability in antibody responses warrants assessment in a sufficient number of patients to characterize variability in antibody responses. Additionally, evaluation of a follow-on NBCD should ensure that anti-drug antibodies do not neutralize drug efficacy or bind to endogenous proteins; and characterize the immunologic effects of switching between a reference NBCD and a generic product.
In many countries generic approval of a follow-on product allows automatic substitution at the pharmacy level. While there is continued pressure worldwide to reduce drug costs, a major concern is whether patient safety and well-being are compromised by automatic substitution or interchange with follow-on products [73]. Commonly, clinicians, caregivers and patients are not aware of the change in medication [11], often to the frustration of prescribing physicians [74–77]. At minimum, substitution of NBCDs without the involvement of a healthcare professional should be discouraged. Generally, patients should not be automatically switched to a generic NBCD if they are doing well. If a switch is unavoidable, the safety and efficacy of the new product should be monitored [78].
In some instances, substituting a lower priced generic for an innovator drug has resulted in higher healthcare utilization and overall costs [16, 17, 25] due to decreased efficacy or adverse events. Overall, drug product replacement that is guided by acquisition cost only may increase other costs and not be cost-effective from the patient’s and payer’s perspective [11].
Conclusion
Patients, physicians, and third-party payers expect generic products to be equally safe and comparably effective to the reference drug. For follow-on NBCDs, this will likely require more thorough assessment than the current generic drug approval process. Ultimately, regulatory requirements for approval and interchangeability of follow-on NBCDs will probably require a ‘case-by-case’ approach.
As FDA approaches the challenge of developing guidelines for follow-on NBCDs, it will be important to include a variety of constituents in the process. Members of the medical community have expressed concerns about the safety and efficacy of biosimilar drugs that indicate an increasing lack of trust of the drug regulatory process, primarily due to ‘an absence of the organized medical community in the public process of creating and updating the guidelines’ [74]. The same may hold true for follow-on NBCDs. Regulators must operate in different worlds to balance legal, scientific, and public health considerations as legislation for approval of follow-on NBCD products evolves. Scientific discussion and multidisciplinary research between experts from academia, industry, the medical community, and regulatory bodies; and consensus discussions with all stakeholders on an international level will aid in development of meaningful regulatory guidelines to ensure the safety and effectiveness of follow-on NBCD products.
Acknowledgement
The author received editorial support from Ms Sheila Truten of MC2, Wynnewood, PA, USA, who does not meet criteria for authorship.
Disclosure of financial and competing interests: Dr J Michael Nicholas is an employee of Teva Pharmaceutical Industries. Teva produces and markets a wide range of generic drug products, as well as the branded glatiramoid, Copaxone.
Provenance and peer review: Commissioned; externally peer reviewed.
References
1. Biologics Price Competition and Innovation Act of 2009, Public Law 111-148, Sec. 7001-7003, 124 Stat. 119. Mar. 23, 2010.
2. U.S. Department of Health and Human Services. Guidance for industry scientific considerations in demonstrating biosimilarity to a reference product. Draft guidance. Februray 2012 [homepage on the Internet]. 2012 Feb [cited 2014 Feb 24]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
3. U.S. Department of Health and Human Services. Guidance for industry biosimilars questions and answers regarding implementation of the Biologics Price Competition Act of 2009. Draft guidance. Februray 2012 [homepage on the Internet]. 2012 Feb [cited 2014 Feb 24]. Available from: http://www.fda.gov/downloads/Drugs/Guidances/UCM273001.pdf
4. U.S. Department of Health and Human Services. Guidance for industry quality considerations in demonstrating biosimilarity to a reference protein product. Draft guidance. Februray 2012 [homepage on the Internet]. 2012 Feb [cited 2014 Feb 24]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf
5. European Medicines Agency. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. EMEA/CHMP/BMWP/14327/2006. 13 December 2007 [homepage on the Internet]. 2008 Jan [2014 Feb 24]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003946.pdf
6. European Medicines Agency. Guideline on similar biological medicinal products. CHMP/437/04. 30 October 2005 [homepage on the Internet]. 2005 Nov [cited 2014 Feb 24]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf
7. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005 Rev. 1. 3 June 2013 [homepage on the Internet]. 2013 Jun [cited 2014 Feb 24]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/06/WC500144124.pdf
8. Nicholas JM. Complex drugs and biologics: scientific and regulatory challenges for follow-on products. Drug Inf J. 2012;46(2):197-206.
9. Schellekens H, Klinger E, Mühlebach S, Brin JF, Storm G, Crommelin DJ. The therapeutic equivalence of complex drugs. Regul Toxicol Pharmacol. 2011;59(1):176-83.
10. Varkony H, Weinstein V, Klinger E, et al. The glatiramoid class of immunomodulator drugs. Expert Opin Pharmacother. 2009;10(4):657-68.
11. Mühlebach S, Vulto A, de Vlieger JSB, Weinstein V, Flühmann B, Shah VP. The authorization of non-biological complex drugs (NBCDs) follow-on versions: specific regulatory and interchangeability rules ahead? Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(4):204-7. doi: 10.5639/gabij.2013.0204.054
12. Woodcock J, Griffin J, Behrman R, et al. The FDA’s assessment of follow-on protein products: a historical perspective. Nat Rev Drug Discov. 2007;6(6):437-42.
13. Toblli JE, Cao G, Oliveri L, Angerosa M. Differences between original intravenous iron sucrose and iron sucrose similar preparations. Arzneimittelforschung. 2009;59(4):176-90.
14. Wadhwa M, Skog AL, Bird C, et al. Immunogenicity of granulocyte-macrophage colony-stimulating factor (GM-CSF) products in patients undergoing combination therapy with GM-CSF. Clin Cancer Res. 1999;5(6):1353-61.
15. Helmers SL, Paradis PE, Manjunath R, et al. Economic burden associated with the use of generic antiepileptic drugs in the United States. Epilepsy Behav. 2010;18:437-44.
16. Labiner DM, Paradis PE, Manjunath R, et al. Generic antiepileptic drugs and associated medical resource utilization in the United States. Neurology. 2010;74(20):1566-74.
17. U.S. Food and Drug Administration. Federal Food, Drug, and Cosmetic Act (FD&C Act). Section 505 of the FDCA (21 USC § 355) [homepage on the Internet]. [cited 2014 Feb 24]. Available from: http://www.fda.gov/regulatoryinformation/legislation/federalfooddrugandcosmeticactFDCAct/default.htm
18. Shah VP, Vulto AG, Muhlebach S. Workshop on the challenges in substitution of nonbiological complex drugs. Am J Health Syst Pharm. 2013;70(21):1879-80.
19. U.S. Food and Drug Administration. FY 14 GDUFA regulatory research priorities [homepage on the Internet]. [cited 2014 Feb 24]. Available from: http://www.fda.gov/Drugs/NewsEvents/ucm367997.htm
20. Regulations.gov. Teva Pharmaceutical Industries Ltd. (Teva Neuroscience, Inc.) – Citizen Petition. Document ID: FDA-2009-P-0555–0001. Docket ID: FDA-2009-P-0555–0001. 13 November 2009. 2009 Nov [cited 2014 Feb 24]. Available from: http://www.regulations.gov/#!documentDetail;D=FDA-2009-P-0555–0001
21. European Medicines Agency. Reflection paper on non-clinical studies for generic nanoparticle iron medicinal product applications. EMA/CHMP/SWP/100094/2011. 17 March 2011 [homepage on the Internet]. 2011 Apr [cited 2014 Feb 24]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/04/WC500105048.pdf
22. Watson DS, Endsley AN, Huang L. Design considerations for liposomal vaccines: influence of formulation parameters on antibody and cell-mediated immune responses to liposome associated antigens. Vaccine. 2012;30(13):2256-72.
23. Cohen BA, Oger J, Gagnon A, Giovannoni G. The implications of immunogenicity for protein-based multiple sclerosis therapies. J Neurol Sci. 2008;275(1-2):7-17.
24. U.S. Department of Health and Human Services. Guidance for industry immunogenicity assessment for therapeutic proteins. Draft guidance. February 2013 [homepage on the Internet]. 2013 Feb [cited 2014 Feb 24]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM338856.pdf
25. Jahn EM, Schneider CK. How to systematically evaluate immunogenicity of therapeutic proteins – regulatory considerations. N Biotechnol. 2009;25(5):280-6.
26. Schellekens H. Factors influencing the immunogenicity of therapeutic proteins. Nephrol Dial Transplant. 2005;20 Suppl 6:vi3-9.
27. Schellekens H, Jiskoot W. Eprex-associated pure red cell aplasia and leachates. Nat Biotechnol. 2006;24(6):613-4.
28. Casadevall N, Eckardt KU, Rossert J. Epoetin-induced autoimmune pure red cell aplasia. J Am Soc Nephrol. 2005;16 Suppl 1:S67-9.
29. Svenningsson A, Dring AM, Fogdell-Hahn A, et al. Fatal neuroinflammation in a case of multiple sclerosis with anti-natalizumab antibodies. Neurology. 2013;80(10):965-7.
30. Sorensen PS, Koch-Henriksen N, Ross C, Clemmesen KM, Bendtzen K; Danish Multiple Sclerosis Study Group. Appearance and disappearance of neutralizing antibodies during interferon-beta therapy. Neurology. 2005;65(1):33-9.
31. Sorensen PS. Neutralising antibodies to interferon-ß – measurement, clinical relevance, and managment. J Neurol. 2006;253(6 Suppl):vi16-vi22.
32. Smith DM, Simon JK, Baker JR Jr. Applications of nanotechnology for immunology. Nat Rev Immunol. 2013;13(8):592-605.
33. Schell RF, Sidone BJ, Caron WP, et al. Meta-analysis of inter-patient pharmacokinetic variability of liposomal and non-liposomal anticancer agents. Nanomedicine. 2014;10(1):109-17.
34. Carstens MG, Camps MG, Henriksen-Lacey M, et al. Effect of vesicle size on tissue localization and immunogenicity of liposomal DNA vaccines. Vaccine. 2011;29(29-30):4761-70.
35. Holloway C, Mueller-Berghaus J, Lima BS, et al. Scientific considerations for complex drugs in light of established and emerging regulatory guidance. Ann N Y Acad Sci. 2012;1276:26-36.
36. Mamidi RN, Weng S, Stellar S, et al. Pharmacokinetics, efficacy and toxicity of different pegylated liposomal doxorubicin formulations in preclinical models: is a conventional bioequivalence approach sufficient to ensure therapeutic equivalence of pegylated liposomal doxorubicin products? Cancer Chemother Pharmacol. 2010;66(6):1173-84.
37. Szebeni J, Muggia F, Gabizon A, Barenholz Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: prediction and prevention. Adv Drug Deliv Rev. 2011;63(12):1020-30.
38. European Medicines Agengy. Reflection paper on the data requirements for intravenous liposomal products developed with reference to an innovator liposomal product. EMEA/CHMP/806058/2009/Rev. 02. 21 February 2013 [homepage on the Internet]. 2013 Mar [cited 2014 Feb 24]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500140351.pdf
39. Gawne-Cain ML, O’Riordan JI, Coles A, Newell B, Thompson AJ, Miller DH. MRI lesion volume measurement in multiple sclerosis and its correlation with disability: a comparison of fast fluid attenuated inversion recovery (fFLAIR) and spin echo sequences. J Neurol Neurosurg Psychiatry. 1998;64(2):197-203.
40. Toblli JE, Cao G, Oliveri L, Angerosa M. Comparison of oxidative stress and inflammation induced by different intravenous iron sucrose similar preparations in a rat model. Inflamm Allergy Drug Targets. 2012;11(1):66-78.
41. Stein J, Dignass A, Chow KU. Clinical case reports raise doubts about the therapeutic equivalence of an iron sucrose similar preparation compared with iron sucrose originator. Curr Med Res Opin. 2012;28(2):241-3.
42. Rottembourg J, Kadri A, Leonard E, Dansaert A, Lafuma A. Do two intravenous iron sucrose preparations have the same efficacy? Nephrol Dial Transplant. 2011;26(10):3262-7.
43. Lee ES, Park BR, Kim JS, Choi GY, Lee JJ, Lee IS. Comparison of adverse event profile of intravenous iron sucrose and iron sucrose similar in postpartum and gynecologic operative patients. Curr Med Res Opin. 2013;29(2):141-7.
44. Mishra A, Dave N, Viradiya K. Fatal anaphylactic reaction to iron sucrose in pregnancy. Indian J Pharmacol. 2013;45(1):93-4.
45. European Medicines Agency. New recommendations to manage risk of allergic reactions with intravenous iron-containing medicines. EMA/377372/2013. 28 June 2013 [homepage on the Internet]. 2013 Jun [cited 2014 Feb 24]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2013/06/WC500144874.pdf
46. U.S. Food and Drug Administration. Draft guidance on iron sucrose. Recommended March 2012; Revised November 2013 [homepage on the Internet]. 2013 Nov [cited 2014 Feb 24]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM297630.pdf
47. Duncan R, Gaspar R. Nanomedicine(s) under the microscope. Mol Pharm. 2011;8(6):2101-41.
48. Brenner T, Arnon R, Sela M, et al. Humoral and cellular immune responses to Copolymer 1 in multiple sclerosis patients treated with Copaxone. J Neuroimmunol. 2001;115(1-2):152-60.
49. Teitelbaum D, Brenner T, Abramsky O, Aharoni R, Sela M, Arnon R. Antibodies to glatiramer acetate do not interfere with its biological functions and therapeutic efficacy. Mult Scler. 2003;9(6):592-9.
50. European Medicines Agency. Reflection paper on nanotechnology-based medicinal products for human use. EMEA/CHMP/79769/2006. 29 June 2006 [homepage on the Internet]. 2006 Jul [cited 2014 Feb 24]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2010/01/WC500069728.pdf
51. Johnson KP. Glatiramer acetate and the glatiramoid class of immunomodulator drugs in multiple sclerosis: an update. Expert Opin Drug Metab Toxicol. 2010;6(5):643-60.
52. Ziemssen T, Schrempf W. Glatiramer acetate: mechanisms of action in multiple sclerosis. Int Rev Neurobiol. 2007;79:537-70.
53. Larché M, Wraith DC. Peptide-based therapeutic vaccines for allergic and autoimmune diseases. Nat Med. 2005;11(4 Suppl):S69-76.
54. Aharoni R, Kayhan B, Eilam R, Sela M, Arnon R. Glatiramer acetate-specific T cells in the brain express T helper 2/3 cytokines and brain-derived neurotrophic factor in situ. Proc Natl Acad Sci U S A. 2003;100(24):14157-62.
55. Aharoni R, Herschkovitz A, Eilam R, et al. Demyelination arrest and remyelination induced by glatiramer acetate treatment of experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105(32):11358-63.
56. Aharoni R, Teitelbaum D, Arnon R, Sela M. Copolymer 1 acts against the immunodominant epitope 82-100 of myelin basic protein by T cell receptor antagonism in addition to major histocompatibility complex blocking. Proc Natl Acad Sci U S A. 1999;96(2):634-9.
57. Kappos L, Comi G, Panitch H, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med. 2000;6(10):1176-82.
58. Bielekova B, Goodwin B, Richert N, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6(10):1167-75.
59. Basile E, Gibbs E, Aziz T, Oger J. During 3 years treatment of primary progressive multiple sclerosis with glatiramer acetate, specific antibodies switch from IgG1 to IgG4. J Neuroimmunol. 2006;177(1-2):161-6.
60. Karussis D, Teitelbaum D, Sicsic C, Brenner T; AC001 multi-center Israeli study group. Long-term treatment of multiple sclerosis with glatiramer acetate: natural history of the subtypes of anti-glatiramer acetate antibodies and their correlation with clinical efficacy. J Neuroimmunol. 2010;220(1-2):125-30.
61. Farina C, Vargas V, Heydari N, Kümpfel T, Meinl E, Hohlfeld R. Treatment with glatiramer acetate induces specific IgG4 antibodies in multiple sclerosis patients. J Neuroimmunol. 2002;123(1-2):188-92.
62. Baumgartner A, Stich O, Rauer S. Anaphylactic reaction after injection of glatiramer acetate (Copaxone®) in patients with relapsing-remitting multiple sclerosis. Eur Neurol. 2011;66(6):368-70.
63. Rauschka H, Farina C, Sator P, Gudek S, Breier F, Schmidbauer M. Severe anaphylactic reaction to glatiramer acetate with specific IgE. Neurology. 2005;64(8):1481-2.
64. Bakshi S, Chalifa-Caspi V, Plaschkes I, Perevozkin I, Gurevich M, Schwartz R. Gene expression analysis reveals functional pathways of glatiramer acetate activation. Expert Opin Ther Targets. 2013;17(4):351-62.
65. U.S. National Institutes of Health. ClinicalTrials.gov. Efficacy and safety of GTR in comparison to Copaxone® (GATE). [cited 2014 Feb 24]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01489254?term=gate&rank=1
66. Gottlieb S. Biosimilars: policy, clinical, and regulatory considerations. Am J Health Syst Pharm. 2008;65(14 Suppl 6):S2-8.
67. Ramot Y, Rosenstock M, Klinger E, Bursztyn D, Nyska A, Shinar DM. Comparative long-term preclinical safety evaluation of two glatiramoid compounds (glatiramer Acetate, Copaxone(R), and TV-5010, protiramer) in rats and monkeys. Toxicol Pathol. 2012;40(1):40-54.
68. Momenta. M356: Generic version of Copaxone [homepage on the Internet]. [updated 2013 Oct 18; cited 2014 Feb 24]. Available from: http://www.momentapharma.com/pipeline/generic.html
69. Momenta. Momenta Pharmaceuticals announces filing of abbreviated new drug application with a paragraph iv certification for generic version of Copaxone [press release]. Cambridge, MA. 2008 Jul 11 [cited 2014 Feb 24]. Available from: http://ir.momentapharma.com/releasedetail.cfm?ReleaseID=320978
70. NATCO collaborates with Mylan for glatiramer acetate. 2008 Jul 11 [cited 2014 Feb 24]. Available from: http://economictimes.indiatimes.com/topic/NATCO-Collaborates-with-Mylan-for-Glatiramer-Acetate
71. Mylan Inc, NATCO Pharma Ltd. To collaborate on worldwide marketing and distribution of generic Copaxone®. 2008 Jun 10 [cited 2014 Feb 24]. Available from: http://www.prnewswire.com/news-releases/mylan-inc-natco-pharma-ltd-to-collaborate-on-worldwide-marketing-and-distribution-of-generic-copaxoner-57430927.html
72. European Medicines Agency. Guideline on clinical investigation of medicinal products for the treatment of multiple sclerosis. CPMP/EWP/561/98 Rev 1. 16 November 2006 [homepage on the Internet]. 2006 Dec [cited 2014 Feb 24]. Available from: http://www.tga.gov.au/pdf/euguide/ewp056198en.pdf
73. Borchard G, Flühmann B, Mühlebach S. Nanoparticle iron medicinal products – requirements for approval of intended copies of non-biological complex drugs (NBCD) and the importance of clinical comparative studies. Regul Toxicol Pharmacol. 2012;64(2):324-8.
74. Ebbers HC, Pieters T, Leufkens HG, Schellekens H. Effective pharmaceutical regulation needs alignment with doctors. Drug Discov Today. 2012;17(3-4):100-3.
75. Lewek P, Kardas P. Generic drugs: the benefits and risks of making the switch. J Fam Pract. 2010;59(11):634-40.
76. Kesselheim AS. The backlash against bioequivalence and the interchangeability of brand-name and generic drugs. CMAJ. 2011;183(12):1350-1.
77. Liow K, Barkley GL, Pollard JR, Harden CL, Bazil CW. Position statement on the coverage of anticonvulsant drugs for the treatment of epilepsy. Neurology. 2007;68(16):1249-50.
78. Schellekens H, Stegemann S, Weinstein V, et al. How to regulate nonbiological complex drugs (NBCD) and their follow-on versions: points to consider. AAPS J. 2014;16(1):15-21.
|
Author J Michael Nicholas, PhD, Vice President, Specialty Life Cycle Initiatives, Global Specialty Medicines, Teva Pharmaceutical Industries, 11100 Nall Avenue, Overland Park, KS 66211, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2014 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


