Manufacture and regulation of cell, tissue and gene therapy products: global perspectives, challenges and next steps
Published on 2022/03/30
Generics and Biosimilars Initiative Journal (GaBI Journal). 2022;11(2):65-80.
Author byline as per print journal: Adjunct Associate Professor Sia Chong Hock, BSc (Pharm), MSc; Christine Koh, BSc (Pharm) (Hon); Associate Professor Chan Lai Wah, BSc (Pharm) (Hon), PhD
|
Abstract: |
Submitted: 29 January 2022 Revised: 11 March 2022; Accepted: 14 March 2022; Published online first: 28 March 2022
Introduction
Cell, tissue and gene therapy products (CTGTPs) are distinct categories of therapeutic products intended for use in humans for curative, prophylactic, palliative or diagnostic purposes [1]. Currently, these revolutionary products target diseases that are managed by therapies with high treatment burdens such as the use of recombinant factor IX protein therapy in lieu of repeated intravenous blood infusions for haemophilia B, or rare genetic diseases where no therapies exist for patients, as in Friedreich’s ataxia FRDA) [2].
Cell therapies include the use of stem cells for regenerative purposes [3], or via the insertion of gene for special receptors such as the chimeric antigen receptor (CAR) onto T-cells, as gene therapies for the treatment of cancers and rare diseases. Gene therapies offer a more permanent solution to genetic diseases, when compared to conventional treatments such as chemotherapy through the introduction of genetic material which produces therapeutic proteins [4]. Typically, this circumvents the restrictions related to the therapeutic use of recombinant peptides, including low bioavailability, clearance rates, and exorbitant production cost [5]. Tissue therapies aim to mend and restore injury to organs and tissues through the engineering of components involving cells and tissue architectures [6], combining cells from a patient with scaffold biomaterials [7], which can potentially plug the severe shortage in donated organs. Currently, there are over a hundred thousand potential recipients on the waiting list in the United States alone [8].
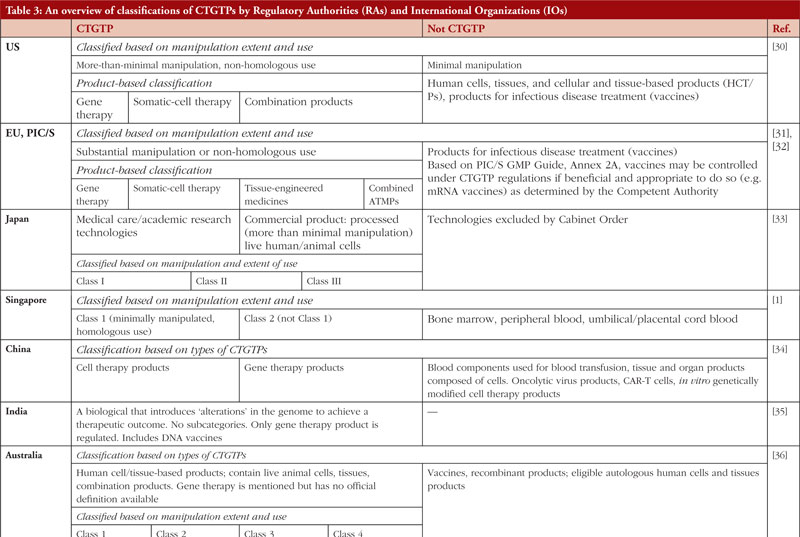
CTGTPs have been defined and classified differently by various regulatory bodies, where a product will go through the regulatory pathway according to the definition that they fall under. In some countries, CTGTPs are also known as advanced therapy medicinal products (ATMPs) or regenerative medicines (RM). In other countries, they may be further classified under categories such as cell therapy medicines, gene therapy medicines, tissue-engineered medicines, or combined products, with different variations in naming the respective product categories. These definitions take into consideration the degree of processing from the starting materials as well as the purpose of the product, such as the restoration of function or prevention of disease, which determines the extent of regulation a product is subject to. However, there are also some countries where little or no regulations for CTGTPs exist.
With rapid progress and strong interest in CTGTPs, as evident from the increase in investments in these novel products over the last decade [7], many challenges have also arisen. These come from a lack of knowledge regarding these novel products, where conventional modes of manufacturing and regulation have not been adequately adapted to ensure the safety and efficacy of CTGTPs. Considering the lack of studies on CTGTPs, this article aims to juxtapose the characteristics of CTGTPs with conventional biologicals, where existing manufacturing trends from the latter will serve as a basis for proposing solutions to solve the challenges faced in manufacturing and regulation. This article also aims to present the manufacturing processes and regulatory frameworks that CTGTPs are subjected to in different countries.
CTGTPs and their principles of action
Cell therapy products use cells to repair or replace injured tissue or cells in the body. Cells used may include mesenchymal stem cells (MSCs), T cells, and pancreatic islet cells [9]. Cell therapy products may contain only cells alone or exist as a part of gene therapy or tissue therapy.
These products can be further categorized according to autologous therapies or allogeneic therapies. Autologous therapies obtain cells directly from the patient, thus circumventing immune reactions. However, this also means that these cells are not suitable for use in mass manufacturing as ‘off-the-shelf’ products. In contrast, allogeneic therapies utilize donated cells to treat multiple patients. These cells are collected from healthy donors, rather than directly from the patient to create a master cell bank (MCB). Consequently, the risk of immunogenic reactions is higher [16]. Products for gene therapies are manufactured via ex vivo or in vivo processes. An in vivo process is one where a viral vector carrying the gene of interest is directly transferred into the body via infusions, whereas an ex vivo process involves removing a patient’s cells to be altered and then re-infusing the genetically modified cells back into the patient’s body [17]. For tissue engineering, current strategies include recreating organ and tissue structure via scaffold fabrication, 3D bioprinting and self-assembly, integration of grafts to host via vascularisation and changing the host environment to create therapeutic responses [6].
Regulators often decide on the stringency of manufacturing requirements by considering if the therapy product is intended for homologous use, where the therapy product is administered at an identical anatomical site and fulfils the same function in the recipient as in the donor [1]. In addition, the extent of manipulation the cells or tissues have undergone is also considered, where minimal manipulation implies that biological traits or functions of the cell or tissue are unchanged [1].
Differences between CTGTPs and conventional biologicals
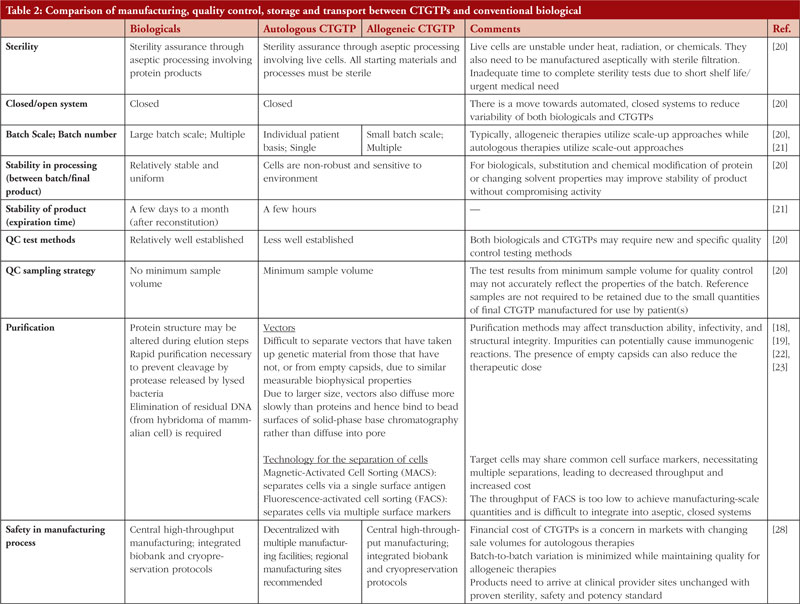
The type of therapy has an immense impact on the type of manufacturing style and challenges encountered. Conventional biologicals are biotherapeutic protein products made with recombinant DNA technology, where cells are reprogrammed genetically to produce proteins that are insufficiently produced in the body [10]. An example includes synthetic insulin for the treatment of diabetes and monoclonal antibodies for cancer treatment. The key difference between conventional biologicals and CTGTPs is that while proteins are the final products in conventional biologicals, cells are the final products in CTGTPs. Cells can produce specific proteins continuously as compared to the fixed number of specific proteins, which may be degraded and thus depleted in the body. Hence, the cells can potentially allow for longer lasting or permanent prophylaxis. In general, the manufacturing process for chimeric antigen receptor T (CAR-T) cell therapy products, one of the most common CTGTPs, involves steps to preserve the cells and focus on purifying specific target cells, while biologicals manufacturing involves an additional step of isolation and purification of the protein. Since cells are relatively more sensitive to their environment, a more stringent process in terms of the manufacturing environment must be in place to maintain the quality and safety of the CTGTP. Overall, cell therapy products are generally not as well characterized as compared to conventional biologicals, thus having different biomarkers in testing for efficacy.
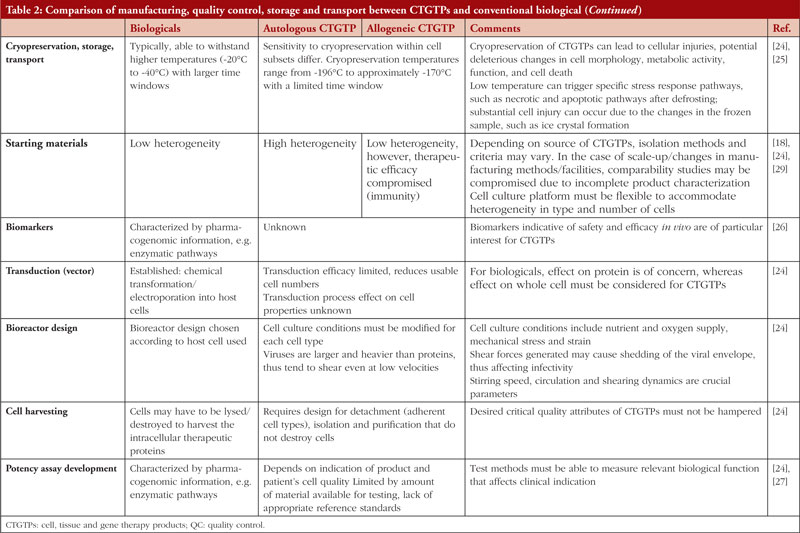
Table 1 shows examples of the different types of products and how they work. Figure 1 focuses on the manufacturing process of CAR-T cell therapy product and conventional biologicals. Table 2 gives further details regarding the manufacturing, quality control storage and transport between CTGTPs and conventional biologicals.


Official definitions of CTGTPs
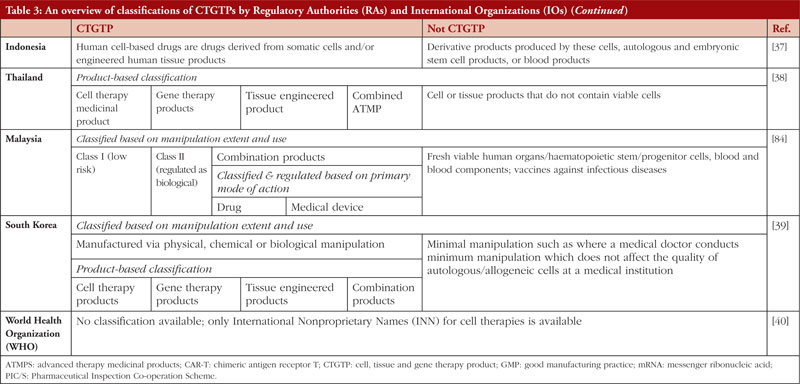
In general, most countries adopt a risk-based approach in deciding if a product is a CTGTP before subjecting it to licensing requirements. In countries such as the United States (US), the European Union (EU), and South Korea, minimally manipulated and homologous use products are not subject to marketing authorization. For CTGTPs which require marketing authorisation, the extent of regulatory requirements depends on the type of products manufactured. Countries such as Singapore include minimally manipulated and homologous products under its definition of CTGTPs, and specifically exclude other products of the same type, e.g. bone marrow, peripheral blood, cord blood and vaccines, without further classifying them as CTGTPs. Other countries such as China and India have no formal definitions for CTGTPs.
Table 3 states the definitions and classifications by the US, EU, Australia, Japan, China, India, Malaysia, Thailand, Singapore, South Korea, the World Health Organization (WHO) and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). These countries have been chosen to represent a heterogeneous regulatory environment with differing levels of capacity and maturity.
Manufacturing of CTGTPs
In the manufacture and commercialization of CTGTP, two main modes of manufacturing styles exist. Decentralized manufacturing spreads production over different locations, allowing for a more amenable response to demands [41]. An example of a decentralized manufacturing process is the integration of good manufacturing practice (GMP) facilities into a hospital setting to produce autologous therapies for patients. However, certain risks are associated with this model, including difficulty in quality control due to a lack of central oversight and contamination risk [42]. On the other hand, centralized manufacturing has been the prevalent way of manufacturing as it enables an economy of scale, due to well-established processes and machinery to produce large, standardized batches of the same product at a single location [41]. Some examples include chemical drugs, allogeneic products and biologicals. However, this characteristic makes it difficult for manufacturers to customize products for specific patients. Currently, centralized manufacturing is utilized for licensed CTGTPs such as Kymriah (Novartis) and Yescarta (Gilead), with a substantial interest in moving towards decentralized manufacturing for autologous products at the point of care [8]. The advantages and disadvantages of the manufacturing methods will be further discussed in the subsequent sections.
As mentioned, the unique nature of CTGTPs as compared to conventional biologicals makes the manufacturing of safe and efficacious CTGTPs a challenge. So far, there has been a clear distinction between manufacturing for product development and conducting clinical trials for academic research, with the former taking place in pharmaceutical companies and the latter in hospitals. However, in recent developments, personalized CTGTPs have demanded a higher involvement of hospitals in the development of CTGTPs due to their manufacturing process [20], which must be near to the patient due to the sensitivity of the product, thus necessitating hospital premises to be of GMP standard. The problems of quality control in cell therapies produced in the academic setting and industry, coupled with strict regulations and difficulty in harmonizing a standardized manufacturing process, hinder the availability of treatments to patients [43] through manufacturing process challenges.
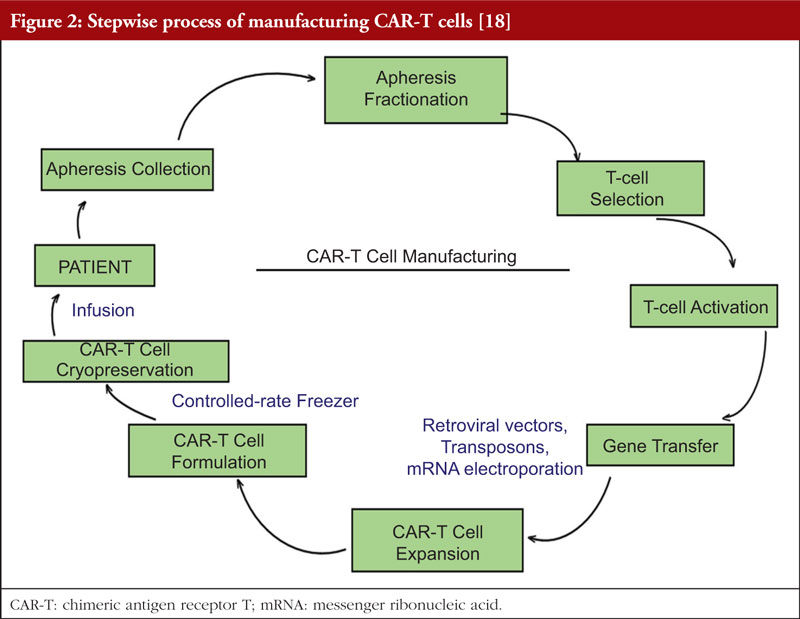
The manufacturing process of CAR-T cells is shown in Figure 2. Peripheral blood mononuclear cells are first collected from the patient via leukapheresis. This is followed by T-cell selection, removing adulterants such as gross red blood cells and platelets, while simultaneously enriching T cells [18]. The T cells are then activated using technologies involving antibody-coated nanobeads before being transduced with a viral vector, which contains the anti-CD19 CAR transgene. The T cells are then expanded in bioreactors to produce doses for sufficient therapeutic effect. Finally, the T cells are removed from the beads, washed, cryopreserved in infusion bags, and tested for the critical quality attributes of the product before it is released and thawed for infusion into the patient [44].
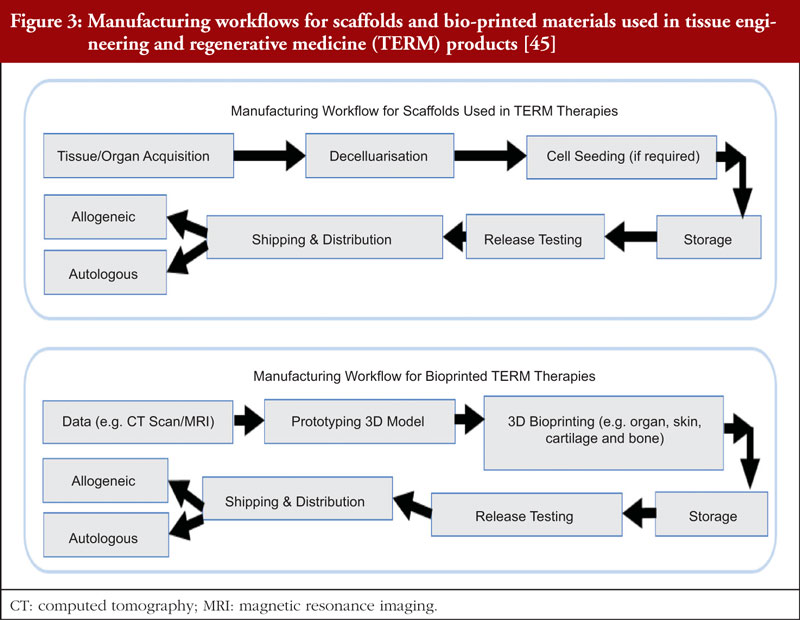
In the case of tissue engineering and regenerative medicine (TERM) therapies, multiple types of materials are required to come together. As such, the manufacturing workflow is different for each component, including cells, scaffolds or bio-printed therapies. The manufacturing workflow for cells is as shown in Figure 2, whilst the manufacturing workflow for scaffolds and bio-printed materials are shown in Figure 3. Bioprinting involves the fabrication of three-dimensional anatomical structures to be used in therapies [45] and in in vitro models since they can be engineered to mimic physiological tissues to a high degree [6]. Other components may include signalling molecules or medical devices [45].
Premises and equipment
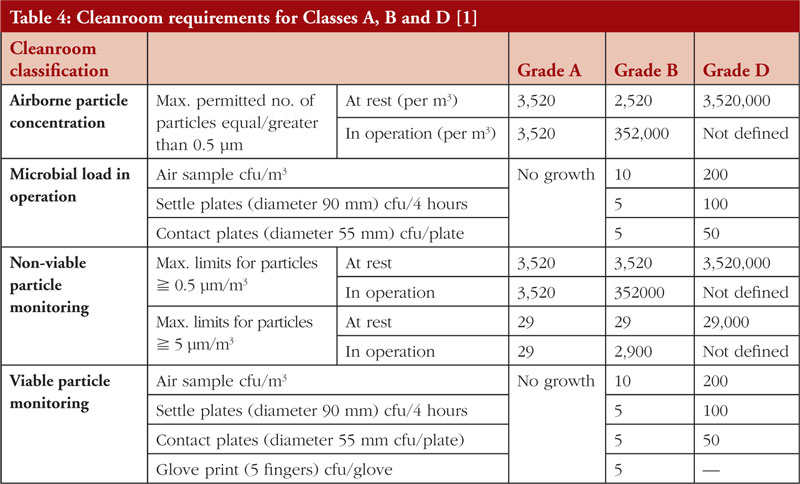
Since CTGTPs cannot be terminally sterilized, they must be aseptically processed as stipulated under national and international CTGTP guidelines, including those of Singapore HSA. This means that a Grade A environment with a Grade B background for open systems, or a Grade D background for closed systems, including isolators, are used to reduce the number of contaminants in an aseptic environment [1]. The relevant cleanroom requirements are listed in Table 4, together with an example of a cleanroom in Figure 4. To reduce the risk of contamination, a closed system is typically preferred, as an open processing system allows for materials to be exposed to the room’s environment. A closed processing system consists of closed tubing pathways and connections, incorporating pre-sterilized, single-use components [46], thus eliminating the introduction of adventitious agents. This allows for flexibility and space in process design for regulatory compliance since materials of closed systems are pre-assembled [47], with closed, automated systems such as the CliniMACS Prodigy from Miltenyi which can perform cell preparation, enrichment, activation, transduction, expansion and purification, allowing for easy integration into manufacturing strategies [48]. A Grade D cleanroom environment also equates to less strict ventilation, cleaning, and gowning requirements [20] which can reduce financial costs. However, due to the novelty of closed process technology, many personnel are still unfamiliar with using the technology, thus preferring the traditional process of biomanufacturing [47]. Additionally, the healthcare technology that is available at hospitals is often not enough to meet all GMP requirements, such as installation, operation and maintenance which require suitably qualified technicians to implement them, often leading to the outsourcing of these activities [48]. Currently, a small number of cell-processing facilities in CTGTP manufacturing are closed systems; however, they are anticipated to increase in the years to come [49].
Materials and processing
Starting material
Since the initial cell populations are obtained from the patients themselves in autologous cell therapies, they can be highly heterogeneous as the quality of the cells depends on the patient’s health. This can reduce the reproducibility and definition of the final product [24]. With a limited amount of starting material available from the patient, the amount of material available for quality control test methods may be correspondingly limited. Thus, the sampling strategies to ensure a sufficient final product dose for patients may in turn affect the accuracy of the test results which reflects the properties of a batch of CTGTP [48]. Several cell types are also adherent in nature, requiring detachment from cultures in their intact form before being processed downstream, thus potentially affecting cell quality [3]. Furthermore, cells are highly sensitive to their environment and display changing behaviours in response, further contributing to their variability [51]. Patients receiving the therapy are also cancer patients who undergo aggressive therapies which can affect T-cell fitness negatively, leading to failure in product generation altogether [52].
Next, as the biophysical properties of full and empty viral capsids are similar, it is a challenge to separate them. This can result in them being processed together, causing the final product to contain both kinds of vectors during the delivery of genetic materials into the cells. If many empty vectors are present, a larger therapeutic dose may be needed, which may lead to adverse immune reactions in patients [53].
For tissue-engineered products, additional materials such as cells, scaffolds and signalling molecules are required to be combined during the manufacture of the product, thus increasing the complexity of the manufacturing process. In addition, there is much uncertainty in terms of whether signalling molecules can bring about the desired cellular differentiation, whether the scaffold supports cellular growth and vascularisation or whether the product has the desired structural properties [7].
Quality control
Due to the limited amount and variability of the material, the traditional approach of process validation can be difficult to perform, especially for autologous products, as it requires multiple successful production batches to be evaluated and potential changes to be made to critical processes, equipment and materials subsequently. Additionally, as the mechanism of actions for cells are not well recognized, the critical quality attributes (CQAs) associated with measurable product attributes for assuring functional quality and reproducibility cannot be determined [54].
The intrinsic variability of cells also contributes to the variability of critical process parameters (CPPs). In the use of cells in tissue engineering, purity is assured since characterization has been achieved for differentiated cells. However, pluripotent cells would require 100% purity to avoid tumorigenicity, thus creating ambiguity in the assessment of their safety and efficacy [55]. Each component, such as the production of scaffolds, would require the maintenance of their quality attributes before seeding them with cells, raising the complexity of tissue-engineered products [45].
The monitoring of CPPs, such as culture conditions, which are elements of the production process that affect CQAs such as cell quality and reproducibility across multiple batches and manufacturing sites have not been established. Extensive data analytics and models would thus have to be used to identify sets of markers (as CQAs) from animal studies and clinical trials to establish product safety [54]. Currently, there is a shortage of robust process analytical technologies (PATs) to examine CPPs for cell-expansion and other processes. Although PAT devices that monitor pH, dissolved oxygen (DO), and other biopharmaceutical CPPs have improved significantly in terms of miniaturization and integration, the manufacture and quality control of CTGTPs are still using monitoring or detection devices that can damage the sample [56]. Overall, CTGTP manufacturing does not currently incorporate quality-by-design principles that allow for high quality and extensive production of therapeutic cells [54].
Cryopreservation
Cell function starts to deteriorate after the collection of the sample and continues up to cryopreservation [25]. In cryopreservation, cells are commonly stored in liquid nitrogen at −196◦C. To recover cryopreserved cells, slow freezing and fast thawing are usually performed [57]. The construction of a cryo-chain for extended storage and delivery is pertinent to preventing the decrease in metabolic activity and cell viability as cells go through different stages in the manufacturing process, which may take up to more than 4 weeks due to quality assessments, release controls and shipment in the case of a centralized process [58]. In contrast, a decentralized process takes up to only 2 weeks as there is no need for cryopreservation nor the shipment of T cells, which protects the cells from injury during freezing and thawing [59]. A shortened duration between leukapheresis and infusion also reduces the need for the patient to receive bridging chemotherapy, which is important in controlling disease progression and averting infections from low blood cell count [58]. A patient would need to receive bridging therapy to control disease progression and prevent potential infections due to low blood cell count while waiting for the infusion [58], thus a shortened duration between leukapheresis and infusion in decentralized processes would also cut down on costs of chemotherapy and extended hospital stay [59].
Human resource and accreditation
Due to the novelty of technology involved in the manufacturing process, human resources would have to be trained, requiring expertise from biology, engineering and computer science [54]. A survey conducted has shown that human resources in multiple fields such as business expansion and product development, together with researchers possessing a comprehensive scope of qualifications, were much sought after. Personnel with GMP manufacturing experiences were also needed by a large portion of respondents [60]. In the context of centralized processing, the efficient allocation of manpower and equipment can be realized [27], if professionals have a thorough understanding of cryopreservation protocols [25], treatment guidelines and possible complications of CTGTP administration, such as cytokine release syndrome and neurotoxicity [8]. Additionally, physicians tend to be clinically conservative and are unwilling to utilize novel treatments especially if they require atypical methods of delivery, thus necessitating evidence in safety assurance and the ability for processes to integrate into existing clinical practice [61, 62].
Potential solutions to the challenges encountered in manufacturing
Outsourcing
Due to the substantial cost of manufacturing CTGTPs, it is important to decide which manufacturing strategy is suitable to cater to a specific product and to design a strong supply chain that can withstand disruptions, such as in a global pandemic where a shortage of materials can result in delayed shipments of cell therapies [63]. One strategy where pharmaceutical start-ups and small developers who lack the resources to manage in-house manufacturing may choose to outsource to contract development and manufacturing companies (CDMO), which are larger and better able to utilize economies of scale [64]. In the absence of trained personnel, it may be prudent to outsource aseptic procedures to avoid potential sterility and contamination issues. If companies struggle to meet guidance standards, they should be strongly encouraged to increase the extent of outsourcing to help in the compliance of GMP standards. It is of critical importance that the CTGTP manufacturing setup complies with GMP standards.
Qualifications
Current GMP guidelines for CTGTPs only require healthcare professionals to have appropriate training in terms of aseptic handling and gowning with no specific qualifications or minimum years of training [1]. While these guidelines cannot be too specific due to the wide range of CTGTPs, higher standards are expected for products that have undergone substantial manipulation as opposed to minimally manipulated products, which are allowed to comply with less stringent standards. Going forward it is recommended that a minimum length of relevant working experience be stipulated for personnel handling CTGTPs. It should also be made mandatory for personnel who are directly engaged in CTGTP processing to undergo training and be certified in basic aseptic techniques. Professionals may also be encouraged to participate in further education programmes to increase their scientific knowledge of CTGTPs. This is particularly important as a lack of skills and knowledge can impact the quality of the product significantly. This can be seen from the trends in biologicals manufacturing where mishandling in production and administration has led to substandard and unsafe protein products [65]. As more products undergo clinical trials, more scientific knowledge will be generated, some of which can then be incorporated into regulations. The requirements for personnel qualifications and training may then be more clearly stipulated based on the types of CTGTP products manufactured.
Technology
A changing mindset is also necessary in terms of embracing emerging trends of Industry 4.0, including disruptive technologies to improve the comparability of products and to reduce contamination. Technologies can be used to model available data in the calculation of operational feasibility and cost implications to avoid unnecessary cost, as shown in a study by Lam C et al. [66], and to improve the quality of products through robust and non-destructive monitoring techniques.
Control of CTGTPs
Current regulatory framework
Table 5 summarizes the regulatory frameworks governing CTGTPs across several jurisdictions. Some are more comprehensive than others in providing for the different types of pathways. Many countries have expedited pathways to cater to the need for CTGTPs to reach patients quickly.
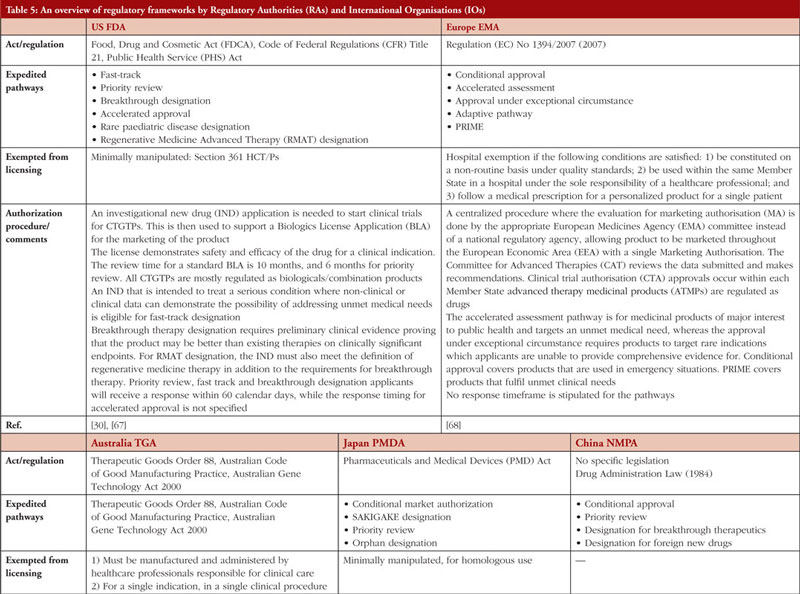
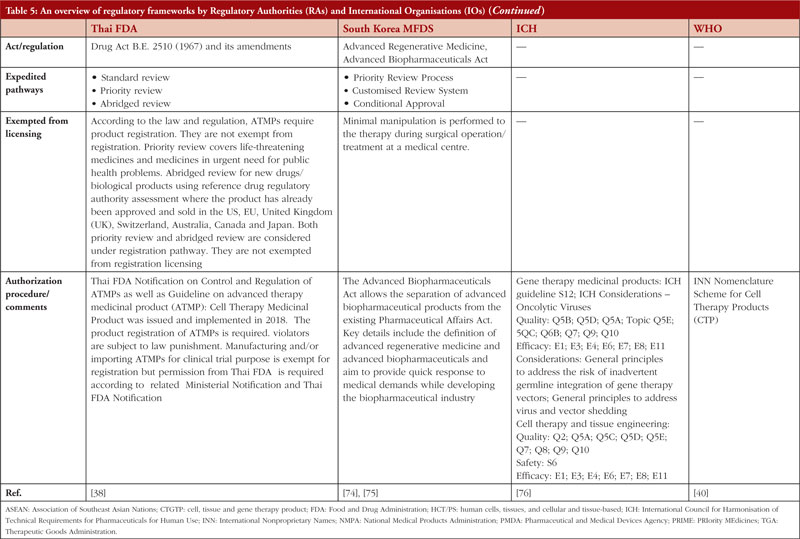
Gaps identified in regulatory control and potential solutions
Risk-based approach
The risk-based approach is contingent upon identifying risks associated with the use of a CTGTP in the clinical setting and its inherent risks concerning quality, safety and efficacy. This process starts at the beginning of product development and matures over time, as the knowledge of the product and its characteristics increases [31]. Expedited pathways shorten this process, allowing therapies to reach patients quickly and increase options available, which is important especially in life-threatening diseases. However, this means that less evidence of safety and efficacy is collected, which may impact product quality [74].
Furthermore, manufacturers have no incentive to perform post-marketing promptly due to potential adverse effects leading to drug withdrawal and subsequently a loss of profit. The post-marketing requirements issued by regulators are also scant on details [72, 77]. Robust post-market evidence is essential in facilitating the successful implementation of accelerated approval programmes for the verification of the safety and efficacy of CTGTPs. However, post-market studies largely utilize methods similar to pre-market trials with minimal patient numbers and brief follow-up periods rather than using real-world scenarios for approved ATMPs in the EU [72, 78]. As such, regulations need to achieve the balance between flexibility in accommodating new knowledge and at the same time specific in terms of setting out requirements in data submission.
In terms of GMP standards, the characteristics of CTGTPs should be considered, such as smaller batch sizes and shorter half-lives. As such, it is important to establish GMP standards different from conventional drugs and biologicals that account for the inherent variability of CTGTPs, where out-of-specification (OOS) products may be anticipated. An example would be the approved CAR-T cell therapy product Kymriah (Novartis), where 10% of products could not be shipped due to OOS issues or manufacturing failures. However, OOS Kymriah is still effective with no evidence of higher safety risk as supported by real-world data [79]. Currently, some countries such as Japan and China have already started to move towards incorporating real-world data into submission requirements, evident from China’s ‘Guideline for the use of Real-World Evidence for Research and Regulatory Review’.
International harmonization of definition and scope of policies
A key discussion in the enactment of Japan’s Act on the Safety of Regenerative Medicine (ASRM) highlighted the importance of shaping the scope of the regulation and deciding what it would regulate, as it could potentially exclude regulation of important developing therapies [80]. The ASRM has also adopted the term ‘regenerative medicine et cetera’, with ‘et cetera’ providing the flexibility to include technologies not yet discovered and unapproved therapies [80]. Discrepancies in regulatory terminologies such as the definition of CTGTPs led to the approval of the same product under different definitions. For example, tisagenlecleucel was approved as a cell therapy product in Australia and Japan yet approved as a gene therapy product in the US and EU [72]. The criteria for eligibility and application procedures for expedited programmes varied across different countries, where no single product was granted expedited approval under the expedited programmes in the US, EU and Japan [72]. This could discourage manufacturers from seeking approval in foreign countries as they would need to spend additional resources to manoeuvre different regulations, thus limiting patients’ access to therapies.
In line with the harmonization of regulations, there is also a need for greater communication between stakeholders. A study showed that only slightly more than half of companies in Malaysia understood the variances in registration requirements between different product classes [83]. Using the example of Kymriah, the US FDA required the product to consist of minimally 80% viable T cells, different from the 70% which was set during clinical trials. However, real-world data revealed that patients receiving doses of viability below 80% also achieved complete response [79]. Thus, there is a need for more extensive communication between different stakeholders, and flexibility in adjusting parameter ranges when more data is received even after the product has been put out to the market.
Furthermore, no global unified regulatory standards are available as a reference or guidance, especially for countries where the technical reviewers lack experience. Also, there are countries which still do not have specific legislation for CTGTPs, including the lack of ethical laws and human rights protection, or having separate regulatory authorities for clinical trial application and marketing authorization [71] unlike the US or UK.
As such, there is a need for the convergence of international regulations. Social science research has shown that emerging markets are stabilized when consumer confidence is boosted through creating standardized regulations [81, 82]. Currently, international harmonization initiatives, including the Gene and Cell Therapy Working Group of the International Pharmaceutical Regulators Form (IPRF) and ICH, are taking place, with the ICH being the primary organization in generating shared technical guidelines to be utilized by different countries in the harmonization process. However, this requires a long time, which may not be rapid enough in dealing with the speed of innovation in the CTGTP area. As such, convergence may be a more desirable option, where regulatory requirements align across countries by utilizing internationally recognized guidance documents [73]. This also provides time for manufacturers to understand any changes to regulations and to adapt manufacturing processes accordingly. By fostering close international cooperation, less advanced countries can also learn from more advanced countries without the need to abruptly adopt regulation standards, since they may not have the capacity to do so currently without being penalized unnecessarily.
Conclusion
CTGTPs are novel therapy products that seek to address unmet medical needs using materials such as stem cells for a regenerative effect, or through genetic engineering to improve the ability of cells to target cancer cells. Types of therapies include autologous therapies, where the starting material is derived from the patient, and allogeneic therapies, where the starting material is obtained from healthy donors. CTGTPs also differ from conventional biologicals in manufacturing process due to their specific characteristics, such as the need to preserve the quality of the entire cell as compared to conventional biologicals, where the focus is on the quality of the therapeutic protein product. Storage, transport and cryopreservation of CTGTPs in the manufacturing cycle are also important since cells require more extreme cryopreservation temperatures, and additional heterogeneity has to be managed if the therapy product is autologous. As a result, a more stringent process is required in the manufacturing of CTGTPs. Some solutions to the challenges encountered in manufacturing include ensuring appropriate accreditation of professionals handling CTGTPs, using automation and outsourcing to CDMOs to ensure better compliance to GMP standards.
In CTGTP regulation, jurisdictions have defined and categorized CTGTPs largely according to the extent of manipulation and whether it is for homologous use or not, where a larger extent of manipulation requires more stringent regulations. Considering the lack of data from clinical trials due to small clinical trial groups and disease severity, many jurisdictions have put in place expedited pathways to allow products to reach patients faster. In addition, regulations need to be flexible to accommodate new scientific knowledge as the CTGTP field advances. The different levels of regulation across countries serve as a barrier to manufacturers seeking to export CTGTPs overseas. In addition, certain countries also do not have specific legislation in place for CTGTPs such as ethical laws and human rights protection. As such, there is a need for the convergence of international regulations to serve as guidance in drafting new laws and to provide clarity to manufacturers. Many guidance documents are already available, such as the ‘PIC/S Annex 2A for Advanced Therapy Medicinal Products’ to guide manufacturers, as new scientific knowledge continues to emerge and evolve. With the help of international organizations, the harmonization of regulations across the world is slowly beginning to materialize, providing guidance and greater clarity to countries that lag on regulations. International harmonization can also minimize the duplication of clinical trials and streamline the process of granting marketing authorizations for CTGTPs, thereby fostering innovation. There should also be greater and more effective communication between the regulator and different stakeholders such as manufacturers, with feedback and some degree of flexibility incorporated into regulations.
The COVID-19 pandemic has accelerated digital transformation and exposed weak areas in pharmaceutical supply chains. At the same time, this has also presented opportunities for manufacturers and regulators to utilize technology to tackle the challenges of CTGTP manufacturing through new and innovative ways, ultimately improving the accessibility of these ground-breaking medicines to patients.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Adjunct Associate Professor Sia Chong Hock, BSc (Pharm), MSc
Christine Koh, BSc (Pharm) (Hons)
Associate Professor Chan Lai Wah, BSc (Pharm) (Hons), PhD
Department of Pharmacy, National University of Singapore, 18 Science Drive 4, Singapore 117543
References
1. Health Sciences Authority. Guidance documents for CTGTP [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.hsa.gov.sg/ctgtp/guidance-documents
2. Tretiakova AP. Realizing the promise of gene therapy through collaboration and partnering: Pfizer’s view [homepage on the Internet]. Scientific American. 2019 Jan 14.
3. Heathman TR, Nienow AW, McCall MJ, Coopman K, Kara B, Hewitt CJ. The translation of cell-based therapies: clinical landscape and manufacturing challenges. Regen Med. 2015;10(1):49-64.
4. Goswami R, Subramanian G, Silayeva L, Newkirk I, Doctor D, Chawla K, et al. Gene therapy leaves a vicious cycle. Front Oncol. 2019;9:297.
5. Belete TM. The current status of gene therapy for the treatment of cancer. BTT. 2021;15:67-77.
6. Chandra PK, Soker S, Atala A. Tissue engineering: current status and future perspectives. In: Principles of Tissue Engineering. Elsevier; 2020. p. 1-35.
7. Gerlovin L, Thompson A, Vailas J. Untapped potential tissue engineering: the three obstacles holding it back. BioProcess Intl. 2021 Apr 26.
8. RESTORE – large scale research initiative in Europe [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.restore-horizon.eu/
9. Association for the Advancement of Blood & Biotherapies. Facts about cellular therapies [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.aabb.org/news-resources/resources/cellular-therapies/facts-about-cellular-therapies
10. World Health Organization. Guidelines on the quality, safety and efficacy of biotherapeutic protein products prepared by recombinant DNA technology, Annex 4, TRS No 987. 18 June 2014 [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.who.int/publications/m/item/recombinant-dna-annex-4-trs-no-987
11. European Medicines Agency. Spherox. 2018 [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/spherox
12. Stein AM, Grupp SA, Levine JE, Laetsch TW, Pulsipher MA, Boyer MW, et al. Tisagenlecleucel model‐based cellular kinetic analysis of chimeric antigen receptor–t cells. CPT Pharmacometrics Syst Pharmacol. 2019;8(5):285-95.
13. Novartis. The science of car-T cell therapy [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.novartis.com/our-focus/cell-and- gene-therapy/car-t/car-t-healthcare-professionals/science-car-t-cell-therapy
14. Cure SMA. Zolgensma [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.curesma.org/zolgensma/
15. About VergenixTM FG [homepage on the Internet]. [cited 2022 Mar 11]. Available from: http://vergenixfg.com/about/
16. Farid SS, Jenkins MJ. Bioprocesses for cell therapies. In: Biopharmaceutical Processing. Elsevier; 2018. p. 899-930.
17. Apte A, Afuwape A, Buljovcic Z, Younes Z. Manufacture and regulation of cell, gene and tissue therapies, Part 1: chemistry, manufacturing, and control challenges. BioProcess Int. 2020.
18. Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015.
19. Sia CH, Sia MK, Chan LW. Global challenges in the manufacture, regulation and international harmonization of GMP and quality standards for biopharmaceuticals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):52-63. doi:10.5639/gabij.2020.0902.010
20. Iancu EM, Kandalaft LE. Challenges and advantages of cell therapy manufacturing under Good Manufacturing Practices within the hospital setting. Curr Opin Biotechnol. 2020;65:233-41.
21. Sterling J. Scaling up cell therapy manufacturing. Genet Eng Biotechnol News. 2018 Sep 7.
22. Morenweiser R. Downstream processing of viral vectors and vaccines. Gene Ther. 2005;12(Suppl 1):S103-10.
23. Murray C, Pao E, Jann A, Park DE, Di Carlo D. Continuous and quantitative purification of T-cell subsets for cell therapy manufacturing using magnetic ratcheting cytometry. SLAS Technol. 2018;23(4):326-37.
24. Roh K-H, Nerem RM, Roy K. Biomanufacturing of therapeutic cells: state of the art, current challenges, and future perspectives. Annu Rev Chem Biomol Eng. 2016;7(1):455-78.
25. Meneghel J, Kilbride P, Morris GJ. Cryopreservation as a key element in the successful delivery of cell-based therapies—a review. Front Med (Lausanne). 2020;7:592242.
26. Schuck RN, Grillo JA. Pharmacogenomic biomarkers: an FDA perspective on utilization in biological product labeling. AAPS J. 2016;18(3):573-7.
27. Quintarelli C, Locatelli F, Caruana I, De Angelis B. Overcoming challenges in car t-cell product CGMP release. Mole Ther. 2016;24(5):845-6.
28. Abou-El-Enein M, Elsanhoury A, Reinke P. Overcoming challenges facing advanced therapies in the EU market. Cell Stem Cell. 2016;19(3):293-7.
29. Mirasol F. New therapies present scaling challenges. BioPharm. 5 Dec 2019.
30. U. S. Food and Drug Administration [homepage on the Internet]. 2021 [cited 2022 Mar 11]. Available from: https://www.fda.gov/home
31. European Medicines Agency [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.ema.europa.eu/en.
32. Pharmaceutical Inspection Co-operation Scheme [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://picscheme.org/
33. Pharmaceuticals and Medical Devices Agency. [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.pmda.go.jp/
34. National Medical Products Administration. Notice of the General Administration on issuing the technical guidelines for the research and evaluation of cell therapy products (2017 No. 216) [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.nmpa.gov.cn/ylqx/ylqxggtg/ylqxzhdyz/20171222145101557.html
35. Government of India. Department of Biotechnology, Ministry of Science & Technology. Stem cells regenerative medicine [cited 2022 Mar 11]. Available from: https://dbtindia.gov.in/schemes-programmes/research-development/medical-biotechnology/stem-cells-regenerative-medicine
36. Australian Government. Department of Health. Therapeutic Goods Administration. Classification of biologicals [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.tga.gov.au/classification-biologicals
37. Jaringan Dokumentasi dan Informasi Hukum Badan Pengawas Obat dan Makanan RI [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://jdih.pom.go.id/
38. FDA Thailand [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.fda.moph.go.th/sites/fda_en/Pages/Main.aspx
39. Korean Legislation Research Institute [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://elaw.klri.re.kr/
40. World Health Organization [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.who.int.
41. Harrison RP, Ruck S, Rafiq QA, Medcalf N. Decentralised manufacturing of cell and gene therapy products: learning from other healthcare sectors. Biotechnol Adv. 2018;36(2):345-57.
42. Medcalf N. Business models for manufacture of cellular therapies. In: Stem cell manufacturing. Elsevier; 2016. p. 265-90.
43. Egri N, Ortiz de Landazuri I, San Bartolomé C, Ortega JR, Español-Rego M, Juan M. CART manufacturing process and reasons for academy-pharma collaboration. Immunol Lett. 2020;217:39-48.
44. Tyagarajan S, Spencer T, Smith J. Optimizing CAR-T cell manufacturing processes during pivotal clinical trials. Mol Ther Methods Clin Dev. 2020;16:136-44.
45. Hunsberger J, Harrysson O, Shirwaiker R, Starly B, Wysk R, Cohen P, et al. Manufacturing road map for tissue engineering and regenerative medicine technologies: manufacturing road map for term. Stem Cells Transl Med. 2015;4(2):130-5.
46. Hampson B. Closed processing for cell therapies. Genet Eng Biotechnol News. 2014;34(9).
47. Paul Mueller Company Academy. Open vs closed bioprocessing [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://academy.paulmueller.com/open-vs.-closed-bioprocessing
48. Viganò M, Giordano R, Lazzari L. Challenges of running a GMP facility for regenerative medicine in a public hospital. Regen Med. 2017;12(7):803-13.
49. Bream A, Salzmann B. The difficulties of manufacturing cell and gene therapies at scale. BioProcess Intl. 2021.
50. Evren A, Pontus B. GMP facilities for manufacturing of advanced therapy medicinal products for clinical trials: an overview for clinical researchers. Curr Gene Ther. 2010;10(6):508-15.
51. Garcia-Aponte OF, Herwig C, Kozma B. Lymphocyte expansion in bioreactors: upgrading adoptive cell therapy. J Biol Eng. 2021;15(1):13.
52. Caldwell KJ, Gottschalk S, Talleur AC. Allogeneic CAR cell therapy—more than a pipe dream. Front Immunol. 2021;11:618427.
53. Mirasol F. Challenges in vector purification for gene therapy. BioPharm Int. 2020;33(2):22-3.
54. Dwarshuis NJ, Parratt K, Santiago-Miranda A, Roy K. Cells as advanced therapeutics: state-of-the-art, challenges, and opportunities in large scale biomanufacturing of high-quality cells for adoptive immunotherapies. Adv Drug Deliv Rev. 2017;114:222-39.
55. Gardner J, Webster A. The social management of biomedical novelty: facilitating translation in regenerative medicine. Soc Sci Med. 2016;156:90-7.
56. Bure K, Lipsitz Y, Lowdell M, Wall I, Zandstra P. Quality by design for advanced therapies: an informed route to enhanced late-stafe clinical success and empowered process flexibility. BioProcess Int. 2020.
57. Nath SC, Harper L, Rancourt DE. Cell-based therapy manufacturing in stirred suspension bioreactor: thoughts for cGMP compliance. Front Bioeng Biotechnol. 2020;8:599674.
58. Buechner J, Kersten MJ, Fuchs M, Salmon F, Jäger U. Chimeric antigen receptor-T cell therapy: practical considerations for implementation in Europe. Hemasphere. 2018;2(1):e18.
59. Ran T, Eichmüller SB, Schmidt P, Schlander M. Cost of decentralized CAR T ‐ cell production in an academic nonprofit setting. Int J Cancer. 2020;147(12):3438-45.
60. Nishigaki F, Ezoe S, Kitajima H, Hata K. Human resource development contributes to the creation of outstanding regenerative medicine products. Regen Ther. 2017;7:17-23.
61. O’Donnell BT, Ives CJ, Mohiuddin OA, Bunnell BA. Beyond the present constraints that prevent a wide spread of tissue engineering and regenerative medicine approaches. Front Bioeng Biotechnol. 2019;7:95.
62. Dlaska CE, Andersson G, Brittberg M, Suedkamp NP, Raschke MJ, Schuetz MA. Clinical translation in tissue engineering—the surgeon’s view. Curr Mol Bio Rep. 2015;1(2):61-70.
63. Qiu T, Wang Y, Liang S, Han R, Toumi M. The impact of COVID-19 on the cell and gene therapies industry: disruptions, opportunities, and future prospects. Drug Discov Today. 2021;26(10):2269-81.
64. Michael L. Manufacturing cures: infrastructure challenges facing cell and gene therapy developers. Pharma Intelligence. In vivo. 2019.
65. Vulto AG, Jaquez OA. The process defines the product: what really matters in biosimilar design and production? Rheumatology (Oxford). 2017;56(Suppl 4):iv14-iv29.
66. Lam C, Meinert E, Yang A, Cui Z. Comparison between centralized and decentralized supply chains of autologous chimeric antigen receptor T-cell therapies: a UK case study based on discrete event simulation. Cytotherapy. 2021;23(5):433-51.
67. Mendicino M, Fan Y, Griffin D, Gunter KC, Nichols K. Current state of U.S. Food and Drug Administration regulation for cellular and gene therapy products: potential cures on the horizon. Cytotherapy. 2019;21(7):699-724.
68. Detela G, Lodge A. EU regulatory pathways for ATMPs: standard, accelerated and adaptive pathways to marketing authorisation. Mol Ther Methods Clin Dev. 2019;13:205-32.
69. Bishop S, Flight S, Thomas N. The regulatory environment for cell therapies in Australia – an opportunity to expedite clinical development. Cell Geni Ther Insights. 2018;4(6):523-33.
70. Australian Government. Department of Health. Therapeutic Goods Administration. Australian regulatory guidelines for biologicals (ARGB) [homepage on the Internet]. 2021 [cited 2022 Mar 11]. Available from: https://www.tga.gov.au/publication/australian-regulatory-guidelines-biologicals-argb
71. Parexel Academy [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://parexel-academy.com
72. Qiu T, Hanna E, Dabbous M, Borislav B, Toumi M. Regenerative medicine regulatory policies: a systematic review and international comparison. Health Policy. 2020;124(7):701-13.
73. Drago D, Foss-Campbell B, Wonnacott K, Barrett D, Ndu A. Global regulatory progress in delivering on the promise of gene therapies for unmet medical needs. Mol Ther Methods Clin Devel. 2021;21:524-9.
74. DIA [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.diaglobal.org/en
75. Korea Biomedicine Industry Association [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://www.kobia.kr/bbs/board.php?tbl=e_newsletter&mode=VIEW&num=72&
76. The International Council for Harmonisation [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://ich.org/
77. Hoekman J, Klamer TT, Mantel‐Teeuwisse AK, Leufkens HGM, De Bruin ML. Characteristics and follow‐up of postmarketing studies of conditionally authorized medicines in the EU. Br J Clin Pharmacol. 2016;82(1):213-26.
78. Fritsche E, Elsallab M, Schaden M, Hey SP, Abou-El-Enein M. Post-marketing safety and efficacy surveillance of cell and gene therapies in the EU: a critical review. Cell & Gene Therapy Insights. 2019;5(11):1505-21.
79. Novartis still hasn’t solved its CAR-T manufacturing issues [homepage on the Internet]. BioPharma Dive. 11 Dec 2019.
80. Takashima K, Morrison M, Minari J. Reflection on the enactment and impact of safety laws for regenerative medicine in Japan. Stem Cell Reports. 2021;16(6):1425-34.
81. Tiwari SS, Raman S, Martin P. Regenerative medicine in India: trends and challenges in innovation and regulation. Regen Med. 2017;12(7):875-85.
82. Charles Edquist [homepage on the Internet]. [cited 2022 Mar 11]. Available from: https://charlesedquist.com/
83. Loh EYX, Goh PS, Mannan AMM, Mohd Sani N, Ab Ghani A. Cell and gene therapy products in Malaysia: a snapshot of the industry’s current regulation preparedness. Cytotherapy. 2021;23(12):1108-13.
|
Author for correspondence: Adjunct Associate Professor Sia Chong Hock, BSc (Pharm), MSc, Department of Pharmacy, National University of Singapore, 18 Science Drive 4, Singapore 117543 |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2022 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


