Payer and physician evidence and discount requirements for biosimilars in three Latin American countries
Published on 2014/12/16
Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(1):11-6.
|
Study objectives: In Latin America, many governments have attempted to address biosimilar safety and efficacy concerns by developing abbreviated regulatory pathways to increase access while controlling quality. This study explores discount and evidence requirements for payers and physicians to provide access to and prescribe biosimilars in Argentina, Brazil and Mexico. |
Submitted: 24 November 2014; Revised: 5 February 2015; Accepted: 9 February 2015; Published online first: 20 February 2015
Study objectives
As with generic small-molecule medicines, the potential for cost savings resulting from the use of biosimilars is attractive to payers worldwide [1, 2]. This attraction has increased sharply in recent years as the impact of biological drugs on health plan budgets has exploded. In Brazil, for example, biotherapeutic products represented 2% of medicines prescribed, yet accounted for 41% of the annual Ministry of Health pharmaceutical budget in 2010 [3]. Biosimilars, however, are different than small molecule generics due to the inherent variability in the production process for biopharmaceutical products and the relatively limited experience that stakeholders have with them. Physicians in particular raise concerns about the degree to which this variability in production may result in differing levels of safety and efficacy of biosimilars relative to their branded equivalents – and each other. Fear of potential immunogenicity issues arising from differences in the biological production process is a concern with biosimilars, and is associated with the need for post-launch pharmacovigilance programmes as seen in Europe. This complex production process is considerably more expensive than that of small molecules, adding to the costs of biosimilars. These factors, along with the matching of prices of biosimilars by originator companies, have resulted in a tempering effect on the launch and uptake of biosimilars as regulators seek to ensure the bioequivalence of biosimilar products through head-to-head demonstration of biosimilarity to their branded originator products.
In Latin America, many governments have attempted to address these concerns by developing abbreviated regulatory pathways for biosimilars in order to increase access while controlling quality [4]. In previous work, we explored the regulatory approaches underway in some of the largest markets in the region [5]. This study builds on that work by exploring the evidence that payers from the selected countries in the region will require to provide access to biosimilars, as well as the price, market access and utilization potential for products that meet these evidence requirements.
Methods
We first conducted online searches, using Google and PubMed, in English, Portuguese and Spanish to update our understanding of the regulatory policies that exist in each of the three target countries: Argentina, Brazil and Mexico. These countries were chosen due to their populations and purchasing power (in terms of gross domestic product per capita) as representative of the most advanced pharmaceutical markets in the Latin American region. Sources included official government websites from each country and key legislation documentation such as Boletín Oficial (Argentina), Diário Oficial da União (Brazil), and Diario Oficial (Mexico). To build on our findings from the literature review, we then conducted exploratory interviews with payers and physician key opinion leaders (KOLs) in each country. Respondents were selected based on referrals from regional industry experts (payers) and PubMed searches (KOLs), and were offered honoraria ranging from US$250 to US$400 for their participation. We are not aware of any potential conflicts of interest among the respondents. Payer informants consisted of two benefits directors in Argentina, one each from a provincial social insurer and a private insurance plan respectively, two advisors to the National Commission for Incorporation of Technologies in SUS (Comissão Nacional de Incorporação de Tecnologias no SUS, CONITEC) and the Ministry of Health in Brazil, and two officials in Mexico, one each from a social insurer and the Ministry of Health respectively. Twenty-five KOLs were emailed an invitation to participate in the research, of which two declined the offer to participate. Physicians interviewed included four KOLs across the three countries, split across two specialties with high levels of biological drug use: haematologist oncologists (two interviews) and rheumatologists (two interviews).
For each of these interviews, we asked a structured set of questions designed to understand what evidence and discount levels payers and physicians will look for when making decisions about providing access to or prescribing biosimilar drugs. Areas of focus included expected impact drivers for the adoption of biosimilars, expected evidence requirements for public access and utilization, payer comfort in providing access versus expected cost savings, and expected discount requirements for access and prescribing. For expected impact drivers, payers and KOLs were asked to rank a list of potential factors in their order of importance in promoting the adoption of biosimilars, where 1 was the least important factor and 7 was the most important factor. For expected evidence requirements for public access and utilization, answers were compiled by collating responses to separate directed question on the topic during interviews into a spreadsheet for subsequent thematic analysis. For payer comfort in providing access versus expected cost savings, payers were asked to specify the opportunity a biosimilar would provide for cost savings to their organization and patients where 1 was no cost savings and 7 was high cost savings. This was repeated for a list of molecules. Then, for each molecule, payers were asked to specify for their level of comfort in promoting the adoption of biosimilars within their organizations where 1 was not at all comfortable and 7 was ‘extremely comfortable’.
Questions focused on a set of drug classes selected as having high potential for the entry of biosimilar products based on secondary research conducted on biosimilar products in development for European, Latin American and the US markets due to the high level of budget impact of the branded biological equivalents of the molecules in these classes. The classes include tumour necrosis factor inhibitors (anti-TNFs), granulocyte colony-stimulating factors (G-CSFs), erythropoiesis-stimulating agents (ESAs) and monoclonal antibodies used in oncology indications (cancer mAbs). Interviews were conducted in Portuguese and Spanish.
Throughout, we have used the term ‘biosimilars’ as the most common usage in the English language literature. However, other terms were noted during our research. For example, biosimilars are frequently referred to as ‘biological products’ in Brazil, as differentiated from ‘new biologicals’ to refer to a new branded agent, ‘biocomparables’ in Mexico, or medicamentos biológicos similares in Argentina.
Results
While biosimilars offer the potential for significant cost savings, safety and quality concerns vary by country, stakeholder segment and therapy area based on a diverse range of factors including regulations, experience with biosimilars and the funding or financial resources of the payer or patient segment in consideration. Findings are summarized below by topic to offer insight into the differences that may exist across the countries included in the research.
Expected impact drivers for the adoption of biosimilars
When considering the attributes of potential therapies that would most impact their decision to adopt biosimilars, payers focus primarily on the total budget impact of the originator product, as well as the clinical value of the product per the evaluation of trusted physician expert advisors. Figure 1 shows the rating of impact drivers in the decision to adopt biosimilars by payers and physicians. This varies somewhat by country, with Brazilian payers placing the highest emphasis on total budget size, perhaps due to the centralized nature of the Brazilian national public healthcare system, the Sistema Único de Saude (SUS). More decentralized markets such as Argentina place a higher emphasis on the level of physician acceptance of the products than on budget impact, particularly in the short-term while clinical value is being proven. Across countries, concern for safety is expressed, particularly in regards to the potential for allergic reactions, but this is not expected to impede adoption as long as safety profiles are considered sufficiently acceptable to gain market approval by regulators. However, payers express skepticism that pharmacovigilance for biosimilars will be as stringent as with innovative biologicals. This theme is more pronounced in Argentina and Mexico than in Brazil, where payers show more concern about providing access to more complex biosimilar molecules such as monoclonal antibodies for more severe conditions. This could potentially reflect the significant budget impact that biological drugs are having on the SUS budget and payers’ increasing interest in biosimilars as a means to provide budgetary relief across disease states. While not one of the top drivers, payers across markets see breadth of indications as an important driver of overall biosimilar adoption. Disease treatment duration (acute versus chronic) and route of administration (intravenous versus subcutaneous) are considered less important factors for payer adoption of biosimilar products across markets.
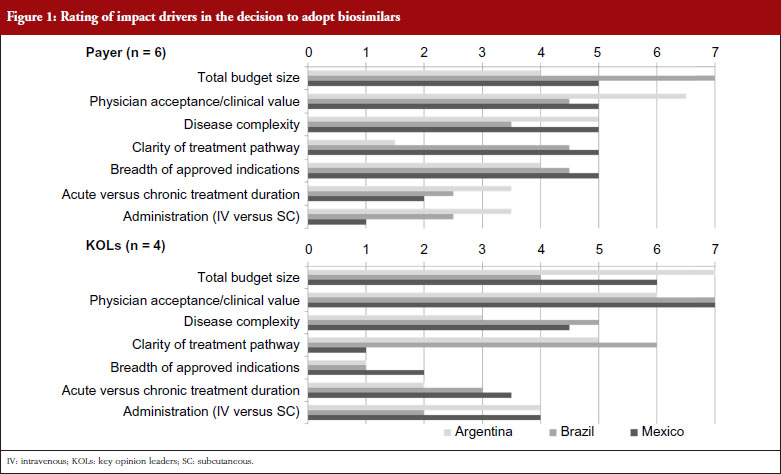
KOLs across markets consistently rate clinical value as the most impactful product attribute to drive the adoption of biosimilar products. Total budget size is seen as having only moderate impact in Brazil, while KOLs in Argentina and Mexico rate budget considerations even higher than their payer countrymen, as they see economic arguments as the principal reason for considering the use of biosimilars, instead of their more established branded originators. Disease complexity is seen as a more important consideration with KOLs than payers in Brazil and Mexico, with KOLs expressing higher levels of concern for the use of more complex molecules in patients with life-threatening conditions, like cancer. Patients with severe and aggressive diseases may not survive long enough to try a different agent if a biosimilar fails to provide the same efficacy benefits, or introduces additional adverse events, compared with the originator molecule. Thus, KOLs are reluctant to use a biosimilar as they perceive that even minor differences in efficacy or safety could have major impacts on treatment outcomes and patient survival. This difference is particularly notable in Brazil, where KOLs worry about the high degree of receptivity to biosimilars that they perceive exists within the SUS. Disease treatment duration (acute versus chronic) and route of administration (intravenous versus subcutaneous) are also considered less important considerations for biosimilar adoption by KOLs, although Argentinian and Mexican KOLs note a moderate level of impact of route of administration due to the increasing preference among rheumatologists and patients for subcutaneous formulations to enable application in the doctor’s office instead of in the infusion clinic. KOLs also state that the number of approved indications does not impact their decision-making, which contrasts with payers, whose primary desire is to ensure that a new biosimilar has the same approved indication as the originator product before permitting access.
Expected evidence requirements for public access and utilization
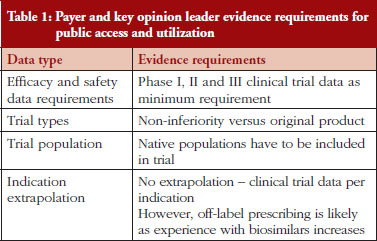
Payers and KOLs consistently report that they will require comparative phase I, II and III trials before providing access to and prescribing biosimilar products, as shown in Table 1, which lists expected evidence requirements to provide public access to and prescribe biosimilar products. This represents a somewhat different viewpoint than those expressed by regulatory experts in previous interviews, who cited the potential for less stringent requirements for less complex molecules such as pegylated interferon and low molecular weight heparin via abbreviated regulatory pathways under development for biosimilars in some countries [4]. Indeed, under the ‘desenvolvimento individual’, or ‘individual development’, pathway in Brazil, ‘non-clinical and clinical studies can be reduced, depending on the amount of knowledge of pharmacological properties, safety and efficacy of the originator product [3]. Thus, Brazil has two regulatory pathways for biosimilars: a more rigorous path for more complex molecules, and a streamlined path for less complex molecules [4]. Payers and KOLs alike state that they will require non-inferior or better bioequivalence in clinical trials to demonstrate that biosimilars have similar efficacy and safety to their branded biological reference molecules. Meanwhile, payers across countries express that they will trust products approved via their countries’ regulatory processes are as safe and effective as their branded equivalents for the indications for which the biosimilars are approved. Similarly, payers state that they will defer the question of indication extrapolation to regulators and reimburse only approved indications, at least in the short-term as patients and clinicians (and payers themselves) gain experience with biosimilars. KOLs are more neutral and state a desire to reserve their opinion on the safety and efficacy of a given biosimilar until they have gained clinical experience with the product.
Payer comfort in providing access versus expected cost savings
Mexican payers stated a relatively low level of comfort permitting access to future biosimilars. This is shown in Figure 2, which depicts payer perceptions of cost savings and comfort providing access to select biosimilars across the drug classes of focus. The payer from the social insurer further clarified that this was due to his perception that, although the Norma enacted by the Mexican Government in September 2012 included regulations pertaining to Good Manufacturing Practices compliance, technical and scientific demonstrating safety, efficacy and quality, and requirements for biocomparability studies and pharmacovigilance of biosimilars [6], specific regulations had not yet been put in place to assess new biosimilar products for bioequivalence. Instead, products were registered and authorized as innovative biotech drugs, although payers cite examples of less complex biosimilars, such as interferon and erythropoietin, that were approved with only comparative data versus their branded reference biological drugs before the Norma was enacted. While payers state that to their knowledge no serious adverse events have been reported, they perceived uncertainty regarding the timing of implementation of bioequivalence assessments in future registration reviews has contributed to the relatively low level of comfort with biosimilars reported by Mexican payers. Furthermore, as a result of examples of more complex biosimilars being approved as innovative biologicals, some biosimilars have launched at prices similar to their branded equivalents, prompting the Mexican payers interviewed to report lower expected cost savings for biosimilars than in the other Latin American markets studied.
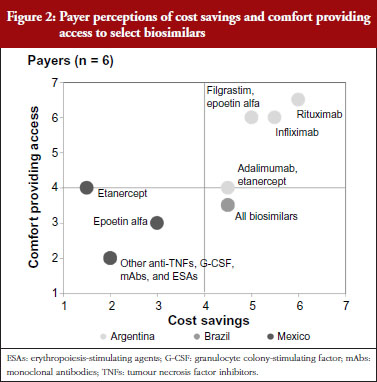
Brazilian payers’ concerns regarding biosimilars are also related to questions regarding assessment processes for regulatory approval. In particular, these payers note concern for products approved via the ‘individual development’ pathway, given the potential to gain approval with less rigorous standards than the ‘comparability’ pathway. Despite these safety concerns, Brazilian payers ultimately state that they will trust the registration decisions made by the Brazilian healthcare regulatory body ANVISA (Agência Nacional de Vigilância Sanitária), and as a result report a moderate level of comfort providing access to biosimilars, with particular interest in high-spend areas like rheumatology, haematology and oncology that have been particularly affected by increases in biological spending in recent years. Given the complexity of biosimilar production, however, payers are also tempered in their price expectations, reporting more moderate levels of anticipated cost savings versus brands than seen with small molecule generics. They note, however, that even relatively modest price discounts would result in significant overall budget impact given the significant biological volume in these drug classes.
Argentinian payers report a higher level of comfort providing access to biosimilars than their Brazilian and Mexican counterparts. This is driven by a higher level of confidence in ANMAT (Administración Nacional de Medicamentos, Alimentos y Tecnología Médica), the Argentinian healthcare regulatory agency, to rigorously assess biosimilar registration applicants for sufficient evidence demonstrating bioequivalent efficacy and safety. This holds for less complex biosimilars as well, as unlike in Brazil, Argentinian payers do not believe that abbreviated regulatory requirements would apply to less complex biosimilar molecules. As for price, Argentinian payers cite higher expected cost savings than their Brazilian and Mexican counterparts due to their belief that lower prices are the primary advantage that biosimilars can offer versus brands. Thus, without substantial cost savings there will be no rationale to justify biosimilar use.
Expected discount requirements for access and prescribing
For biosimilars that meet the evidence requirements discussed earlier and that gain regulatory approval, Brazilian payers report that a 15–30% discount below the price of the branded originator would be necessary for them to provide access to the biosimilar along with the brand, as shown in Figure 3. Since SUS purchases are made at the molecule level, usually to the lowest bidder, the discount levels to win SUS tender contracts could potentially be even lower in the future as biosimilars gain more experience in the market and competitive forces drive the originator price down. However, as the naming system for biosimilar mAbs has not yet been well defined by ANVISA, it is unclear if clinicians will be able to specify originators over biosimilars for SUS patients as they can in the private market. Based on their experience with the private market, payers believe discounts greater than 35% below the branded originator could start to prompt private payers to require patients to first try a biosimilar before reimbursing use of its branded equivalent, or requiring the patient to pay the difference in cost between the biosimilar and branded biological to use the brand first.
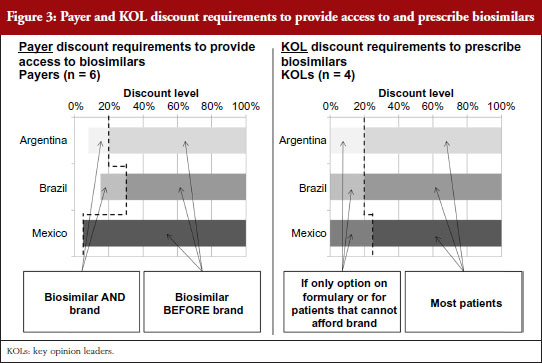
Argentinian payers respond similarly, citing a 10–20% discount below the branded price to provide access to the biosimilar as well as the brand. At discounts greater than 20% they report that they would attempt to restrict access to the brand by asking physicians in their networks to prescribe by molecule name without mentioning the brand or by excluding the original brands from pharmaceutical supply agreements if brand prices and payment terms varied significantly from those for their biosimilar counterparts.
In Mexico, because the healthcare regulatory body COFEPRIS (Comisión Federal para la Protección contra Riesgos Sanitarios, Federal Commission for the Protection against Sanitary Risks) registers biosimilars under the same product codes as their reference brands, social insurers like the Mexican Institute of Social Security (Instituto Mexicano del Seguro Social, IMSS) and the State Employees’ Social Security and Social Services Institute (Instituto de Seguridad y Servicios Sociales de los Trabajadores del Estado, ISSSTE) cannot differentiate between them. As a result, biosimilars compete on equal footing as their reference brands at these public institutions via reverse auction tenders – tender offerings awarded to the manufacturer offering the lowest price determined by a downward-moving auction format – which usually result in discounts of as little as 5% being sufficient to purchase one biological product over another.
Across countries, KOLs cite a strong preference to prescribe branded biologicals over biosimilars due to their proven track records over years of study and market experience. However, KOLs across countries consistently acknowledge that payer access restrictions or patient affordability limitations could cause them to prescribe biosimilars instead of brands at discounts lower than 20–25%. Once the safety and efficacy of a given biosimilar is proven in the market, KOLs state that they likely will consider biosimilars for all patients at discount levels greater than 20-25%.
Conclusion
When asked what attributes of potential therapies would most impact their decision to adopt biosimilars, the small sample of Latin American payers from the markets included in our research focused primarily on the total budget impact of the branded equivalent of the product, as well as the clinical value of the product from respected clinical advisors. Similarly, Latin American KOLs across the selected countries consistently rated clinical value as the most impactful product attribute to drive the adoption of biosimilar products, and saw budgetary concerns as a secondary but still important consideration. Indeed, KOLs in Argentina and Mexico rated budget considerations even higher than their payer countrymen, as they saw economic considerations as the only rationale for considering the use of biosimilars instead of their more established branded equivalents.
While all respondents reported that they will require the full range of comparative trials before accepting biosimilar products, payers across markets consistently stated that they will defer to the regulatory authorities in their respective markets to determine whether biosimilars are safe and effective, and therefore felt comfortable providing access for approved indications. Payers expressed hesitation to provide access for extrapolated indications that have not been granted approval by the regulatory authorities, i.e. off-label use, at least in the short-term while more experience is gained with new biosimilar products. KOLs not surprisingly were more concerned with clinical considerations, and in most cases will only consider use of a given biosimilar for an extrapolated indication, regardless of their regulatory status, once they are confident of its safety and efficacy in the indications where direct clinical evidence support exists. KOL sensitivity to the issue of indication extrapolation, indeed, can also vary by specialty of the clinician. This topic has been brought to the fore since the European Medicines Agency’s decision in September 2013 to grant infliximab biosimilars Remsima and Inflectra authorization for use in the same indications as their originator Remicade [7]. These included the gastroenterological conditions Crohn’s disease and ulcerative colitis, based on extrapolation of rheumatoid arthritis and ankylosing spondylitis clinical trial data (phase 3 and phase 2, respectively). Conversely, Health Canada subsequently approved the infliximab biosimilars for use in all of the originator’s indications except Crohn’s disease and ulcerative colitis [8]. As a result, gastronenterologists may well have had different perspectives on the topic of indication extrapolation than the rheumatologists and haematologist oncologists interviewed as part of this research.
While payers’ range of comfort in providing access to biosimilars varied by market, within the respective markets payers were more comfortable providing access to biosimilars of less complex products like epoetin alfa, or products with which they already had experience with a biosimilar version, while expressing lower levels of comfort in providing access to more complex biosimilar monoclonal antibodies. Payers’ expected cost savings from biosimilars also varied by market, and reflected the extent to which they expected regulators to put in place specific mechanisms to assess new biosimilar products for bioequivalence. As a result, Mexican payers cited lower expected cost savings than their Argentinian counterparts, who expected ANMAT to enact appropriate measures to enable biosimilars to appropriately demonstrate bioequivalence without incurring the full cost associated with the clinical programme of a novel biological.
As for expected discount requirement, payers’ responses ranged from as low as 5% in Mexico, where biosimilars at social insurers like IMSS and ISSSTE compete on equal footing as their reference brands in reverse auction tenders that include both branded and biosimilar competitors, to 10–30% in Brazil and Argentina. Discount levels to win public SUS tender contracts in Brazil, which like public payer tenders in Mexico are conducted at the molecule level, could potentially be even lower in the future as biosimilars gain more experience in the market. At discounts below the branded originator greater than 20% in Argentina and 35% in Brazil, payers reported that, based on their experience with the private market, private payers would start to implement measures to require patients to first try a biosimilar before reimbursing use of its branded equivalent or else pay the difference in cost between the biosimilar and branded biological to use the brand first. While KOLs across markets expressed a strong preference to prescribe branded biologicals over biosimilars, they consistently noted that payer access restrictions or patient affordability limitations could cause them to prescribe biosimilars instead of branded biologicals at discounts less than 20–25%. They went on to note that once they become comfortable that biosimilars indeed have bioequivalent safety and efficacy that they likely will consider them first for all patients at discount levels greater than 20–25%. This finding was consistent across the different countries included in this research despite significant differences in payer concentration, funding and public versus private payer membership seen across their respective healthcare systems.
Our study is subject to some limitations, including the small number of interviews. Because payer respondents were from different payer systems in the countries studied, they may have had different expectations regarding the entry of biosimilars in their regional market. For example, Brazilian payers were advisors primarily for the public system, which provides the majority of biologicals, while Argentinian payers were from a provincial and private insurer, representing multiple payer systems. These differences may affect the interpretation of our findings.
In conclusion, while legitimate questions exist regarding the safety and efficacy of biosimilars, our small, selected sample of payers and physicians across Latin American markets noted the potential for biosimilars to provide cost savings for patients and healthcare systems. Although payers and physicians alike cited the importance that bioequivalent safety and efficacy be proven through head-to-head demonstration of biosimilarity to branded originator products, they ultimately will look to regulators for guidance on which products have provided sufficient evidence, and for which indications. While the level of discount versus the branded originator required for public sector access varied by market, once proven and if offered at discounts greater than 20–25% below their originators, biosimilars have the potential to gain broad penetration not only with cost-sensitive public payers but also with clinically-oriented physicians across Latin American markets.
Disclosure of financial interests: Mr Erik Sandorff, Dr André Vidal Pinheiro and Ms Daniele Severi Bruni are employees of ICON Commercialisation Strategy & Science, an ICON plc company that provides consulting services to biopharmaceutical clients. No client sponsorship was involved with this project. Dr Ronald J Halbert was an employee of ICON Commercialisation Strategy & Science during the time the work was done. Adjunct Professor Valderilio Feijó Azevedo participates in Pfizer and Merck Serono’s international advisory boards for biosimilars. He declares not to have received any payment to collaborate on this project. ICON Commercialisation Strategy & Science provided financial support to this project.
Competing interest: The authors have indicated that they have no conflicts of interest with regard to the content of this article.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Erik Sandorff1, MA, MBA
André Vidal Pinheiro1, PhD
Daniele Severi Bruni1, MPhil
Ronald J Halbert2, MD, MPH
Adjunct Professor Valderilio Feijó Azevedo3,4, MD, PhD, MBA
1ICON Commercialisation Strategy & Science, North Wales, PA 19454, USA
2University of California, Los Angeles (UCLA), School of Public Health, Los Angeles, CA 90095, USA
3Department of Internal Medicine, Hospital de Clínicas, Federal University of Paraná, 224 Rua Alvaro Alvin, Casa 18 Seminário, Curitiba-Paraná PR 80740-260, Brazil
4Edumed Health and Educational Research, Curitiba, Brazil
References
1. Befrits G. The case for biosimilars–a payer’s perspective. Generics and Biosimilars Initiative Journal (GaBI Journal). 2013;2(1):12. doi:10.5639/gabij.2013.0201.009
2. Höer H, de Millas C, Häussler B, Haustein R. Saving money in the European healthcare systems with biosimilars. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(3-4):120-6. doi:10.5639/gabij.2012.0103-4.036
3. Castanheira LG, Barbano DB, Rech N. Current development in regulation of similar biotherapeutic products in Brazil. Biologicals. 2011;39(5):308-11.
4. Kirchlechner T. Current regulations of biosimilars in the Latin American region. Paper presented at: 3rd Annual Drug Information Association (DIA) Latin American Regulatory Conference; 12–15 April 2011; Panama City, Panama.
5. Azevedo VF, Sandorff E, Siemak B, Halbert RJ. Potential regulatory and commercial environment for biosimilars in Latin America. Value in Health Regional Issues. 2012;1(2):228-34.
6. SEGOB. NORMA Oficial Mexicana de Emergencia NOM-EM-001-SSA1-2012, Medicamentos biotecnológicos y sus biofármacos. Buenas prácticas de fabricación. Características técnicas y científicas que deben cumplir éstos para demostrar su seguridad, eficacia y calidad. Etiquetado. Requisitos para realizar los estudios de biocomparabilidad y farmacovigilancia. 2012 Sep 20 [homepage on the Internet]. [cited 2015 Feb 5]. Available from: http://dof.gob.mx/nota_detalle.php?codigo=5269530&fecha=20/09/2012
7. European Medicines Agency. Inflectra. EMA/402688/2013 EMEA/H/C/002778 [homepage on the Internet]. 2013 Jul 19 [cited 2015 Feb 5]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002778/human_med_001677.jsp&mid=WC0b01ac058001d124
8. Health Canada. Inflectra. DIN: 02419475 002778 [homepage on the Internet]. 2015 [cited 2015 Feb 5]. Available from: http://webprod5.hc-sc.gc.ca/dpd-bdpp/info.do?code=90410&lang=eng
|
Author for correspondence: Erik Sandorff , MA, MBA, 2100 Pennbrook Parkway, North Wales, PA 19454, USA |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


