Quality assessment of biosimilars in Colombia – reducing knowledge gaps
Published on 2018/01/19
Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(2):79-83.
|
Abstract: |
Submitted: 7 February 2018; Revised: 8 May 2018; Accepted: 9 May 2018; Published online first: 22 May 2018
Introduction
Similar Biotherapeutic Products (SBPs or biosimilars) have emerged as a new class of biotherapeutic agent. They are being increasingly developed and are becoming more available worldwide. However, approaches to how they are regulated vary across the world. The European Union (EU) has successfully pioneered regulatory procedures and has over 10 years of experience in the regulation and approval of biosimilars. In light of this, other countries across the globe are turning to regulatory experts from Europe and the US for advice on how best to conduct quality assessment of biologicals/biosimilars, to ensure that safety and efficacy of treatment is upheld.
To facilitate discussion concerning quality assessment of biologicals/biosimilars in Colombia, the Generics and Biosimilars Initiative (GaBI) organized an educational workshop [1] and a follow-up meeting [2] in collaboration with the National Food and Drug Surveillance Institute of Colombia (Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA), in 2016 and 2017 respectively. Presentations were made by both international speakers, as well as regulators from Colombia.
The First INVIMA Educational Workshop on the Assessment of Similar Biotherapeutic Products [1] was an interactive event held on 14 June 2016. The format used was similar to that used in prior educational workshops as reported in GaBI Journal [3, 4]. More details of the methods and case presentations can be found in the published reports of the First Latin American Educational Workshop on Similar Biotherapeutic Products [3] and the First MENA Educational Workshop on Regulation and Approval of Similar Biotherapeutic Products/Biosimilars [4]. Summaries of the presentations that were given on analytical comparability, clinical and non-clinical assessment, and safety assessment, are available in the published reports of the Roundtable on biosimilars with European regulators and medical societies [5].
The Second Colombian Scientific Meeting on Quality Assessment of Biosimilars/Similar Biotherapeutic Products [2] was held on 15 August 2017, and focussed on the key topics of: comparability of production processes, design and execution of analytical comparability studies/forced degradation studies, analytical methods, preparation and production of reference standards, and development and validation of host cell protein assays, with a keynote presentation on the Norwegian NOR-SWITCH study on the replacement from originator product to biosimilar infliximab. The meeting was chaired by Dr Elaine Gray and Dr Paul Matejtschuk, who are both Principal Scientists at the UK’s National Institute for Biological Standards and Control (NIBSC).
Both Colombian meetings were held in Bogotá. The list of speakers and the slides they presented are available on the GaBI Journal website (www.gabi-journal.net/about-gabi/educational-workshops).
2016 Quality Assessment of Biosimilars Educational Workshop
In addition to the expert presentations as delivered in prior meetings [3–5], Dr Elwyn Griffiths, former Director General of the Biologics and Genetic Therapies Directorate, Health Canada, gave a presentation entitled ‘Regulatory assessment of already approved rDNA-derived biotherapeutics’, which was an update to that given at the First Turkish Interactive Workshop on Regulation and Approval of Similar Biotherapeutic Products/Biosimilars [4]. He highlighted the role of the World Health Organization (WHO) to ensure global harmonization and regulation of rDNA-derived biotherapeutics. He also summarized the issues they have encountered in implementation, particularly related to products already approved prior to the development of regulatory processes. He introduced the WHO’s new guideline for stepwise product specific regulatory assessment and highlighted the WHO document on regulatory assessment on rDNA derived biotherapeutics.
Summary of the discussion that followed the regulatory presentations of the 2016 workshop
Initially, a query was raised on whether clinical data is required when a manufacturing process is scaled up; with a simple linear up-scaling of a process it was considered that clinical data is not generally required but if there are procedural changes, then such data may be required and comparability cannot be assumed. Currently, for a biosimilar to be approved, clinical data must be supplied to support biosimilarity.
When asked if small biologicals approved without clinical studies, could be termed biosimilars, it was noted that this had not been done in Europe nor in the US. It was explained that, in biosimilar production, a biosimilar does not need to be manufactured using the same expression system as the reference biological and, in practice, using the exact same system would be impossible. However, the manufacturing process must produce an active substance sufficiently similar to that produced by the originator manufacturer. When developing a biosimilar, companies create a database based on a relatively large number of reference batches in order to have sufficient information on the reference product to produce their own Quality Target Product Profile (QTPP). This is not directly required by regulators, but such data is needed to ensure product comparability.
Regarding compiling comparability data, the biosimilar manufacturer must also produce data on the reference product at the same time as producing data on the biosimilar. This data is not readily available and, although some results relating to originator products are published on occasion, these should always be reproduced and made available by the biosimilar manufacturer in order for them to undertake comparability studies. For clinical comparison, pharmacokinetic (PK) and pharmacodynamic (PD) comparisons are a minimum requirement for authorization of biosimilars and in addition, a clinical assessment of the immunogenicity.
Parallel case study working sessions
Case study working sessions took place following the presentations (downloadable from the GaBI website [1]). These followed the same format as those described in previous workshops [3, 4, 6], where two fictional cases of follow-on biological therapeutics are described, one an erythropoietin product and the other a monoclonal antibody. The participants were divided into discussion groups where they evaluated the fictional data supplied. The group discussions are summarized in Tables 1 and 2.
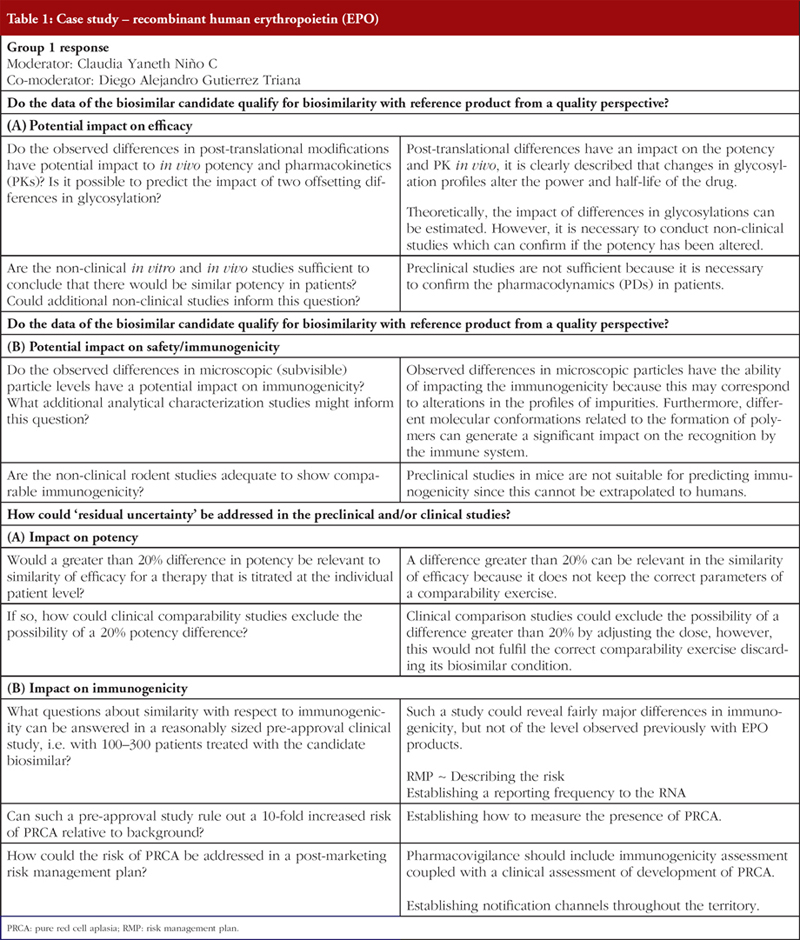
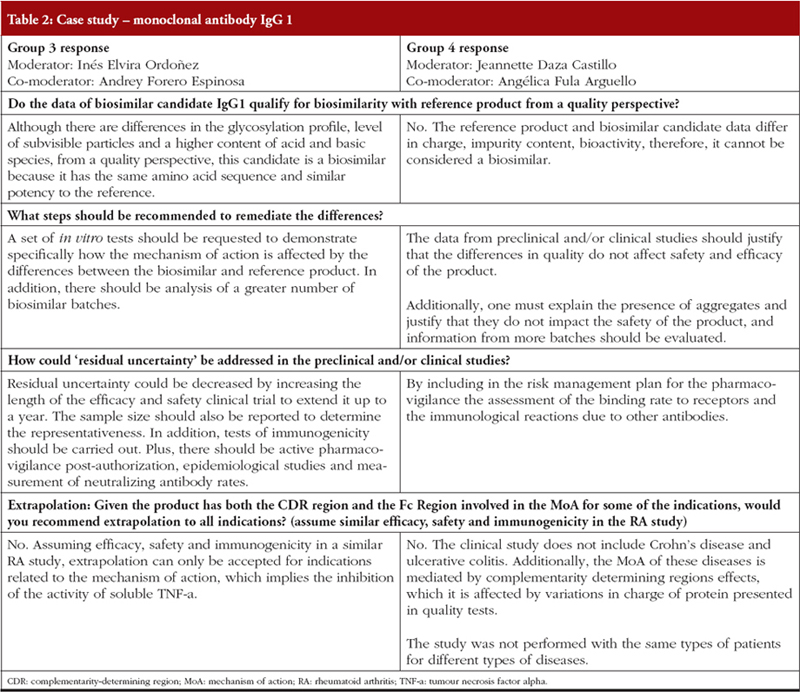
2017 Quality Assessment of Biosimilars/Similar Biotherapeutic Products Scientific Meeting
The 2017 meeting began with Welcoming Remarks by Mr Javier Humberto Guzmán Cruz, Director General of INVIMA, who gave a short presentation on the current development of INVIMA and the health system in Colombia.
The presentation began with an outline of the progress made in Colombia in the last 25 years in terms of universal health coverage. Mr Guzmán noted that Colombia is now in the process of entering the Organisation for Economic Co-operation and Development (OECD). As part of this, experts visited Colombia and carried out a review of Colombia’s health system in which they stated that, ‘Colombia has a well-designed health system, with broadly effective policies and institutions that other countries could learn from and that deserves to be better known internationally’. However, he added that the health system in Colombia is fragile and needs to remain sustainable. To ensure this, pharmaceutical policy needs to be kept up-to-date and innovations incorporated in a controlled manner to guarantee access to safe and efficacious products. Here, Mr Guzmán noted that INVIMA can have an important role in influencing pharmaceutical policy.
INVIMA was created in 1993, and the institute became officially established in 1995, following the passing of a law that aimed to achieve healthcare reform. It has grown substantially in 25 years and now has a budget of US$48 million and many facilities. However, INVIMA faces challenges in the coming years, particularly those posed by the entry of products from large pharmaceutical manufacturers to the Colombian market. Regulators and the Colombian health system need to do work on deciding what gets included in the market to enable the development of frameworks that encourage competition to create access to generics and biosimilars. Mr Guzmán stated that, in Colombia, improving competition that guarantees quality, safety and efficacy is more important than regulation of the pricing of therapeutics. In the last seven years, no biosimilars have been approved, as the guidelines for regulatory approval were not yet developed. Now that the regulations on biologicals/biosimilars are in place, the next challenge is effective implementation. INVIMA has the role of implementing the new regulation on biologicals/biosimilars, and staff are trained both internally and abroad to implement this new legislation. Nonetheless, they are keen to receive advice from those in Europe and the US, who have useful experience in implementation of similar regulations.
Following the Welcoming Remarks, a series of additional presentations were given by experts from Europe and the US (presentations downloadable from the GaBI website [2]). A full manuscript on the presentation of ‘Biosimilar regulations in Colombia,’ is published in GaBI Journal [7]; and information about the presentation entitled ‘Switching from originator products to biosimilars in rheumatology, dermatology and gastroenterology: clinical evidence,’ which described the NOR-SWITCH study, is also published in GaBI Journal [8].
Summary of the discussion that followed the speaker presentations of the 2017 meeting
Discussion following ‘Biosimilar regulations in Colombia’
Dr Gray asked if Colombia expects that a national pharmaceutical company would attempt to register for the three different pathways for products in the future, either via the complete dossier, comparability, and abbreviated comparability approach. Ms Garcia responded that currently, Colombia does not yet have laboratories that will submit information to comply with the new regulations as they have not yet been implemented, however, there are interests from multinational companies to support this requirement.
Discussion following ‘Switching from originator products to biosimilars in rheumatology, dermatology and gastroenterology: clinical evidence’
Professor Tore Kristian Kvien was asked about the results obtained in the switching clinical trials in Scandinavian populations and to what extent these results can be extrapolated to populations, such as the Colombian population. He stated that, in terms of efficacy, there should be no concern using the data with another population and that the NOR-SWITCH data can be used in Colombia.
With regard to patients whose treatment was switched to the Remsima biosimilar and whose disease then worsened, Professor Kvien noted that all patients were followed for one year, and those whose condition worsened also received additional treatment. If they had progression of their disease that required some change in therapy, then they were switched to another biological agent and no patients received the originator Remicade.
In relation to pharmacovigilance data, Professor Kvien noted that this was not part of the NOR-SWITCH study because it was a randomized clinical trial. However, he added that, in Norway, there is a registry called the NOR-DMARD, the Norwegian Antirheumatic Drug Register, and that the data from NOR-DMARD was also examined regarding patients who started with the biosimilar infliximab and patients who have switched from Remicade to Remsima. There does not seem to be any major concern with regards to differences in pharmacovigilance data. Despite this, he noted that the data from NOR-DMARD are not as robust as the data from DANBIO (the nationwide registry for biological therapies in Denmark), which includes some pharmacovigilance data which also support switching.
When asked if he thinks clinical trials are absolutely necessary to confirm efficacy and safety of a biosimilar agent, Professor Kvien said there is a general agreement that, with the current regulations, preclinical quality data need to be generated which support biosimilarity, but this needs confirmation using clinical studies (as outlined in his presentation for CT-P13 and SB4) [8]. Regarding whether or not a NOR-SWITCH study for every biosimilar is needed, another randomized switch study may be required. He gave the example of adalimumab, which is in need of this as it is also immunogenetic. However, it may not be feasible to perform a blinded controlled study of switching from an originator to a biosimilar self-administered drug.
Given the progress in Norway with the adoption of biosimilars, Professor Kvien noted that they would continue on the current path and introduce more biosimilars as they enter the market. The annual tender system means that the system is very competitive which leads to lower prices for the different biosimilars, and the originator products. Regarding his advice for Colombia, he thinks that in general, transparency and competition is important. It is also important that regulators collaborate with key opinion leaders and experts in the different clinical disciplines, so that there are alliances with the clinical environment, which will help to implement the use of biosimilars in Colombia.
Discussion following ‘Preparation and production of reference standards in support of biotechnology products’
Dr Matejtschuk was asked about secondary standards and what they are used for. He said that a secondary standard might be a pharmacopoeial standard and gave the example of a standard from the United States Pharmacopeia (USP) or European Directorate for the Quality of Medicines & HealthCare (EDQM). This will not usually be a primary standard as the dose is not defined, but will actually be a secondary reference material. He noted that working standards could also be secondary standards. Dr Gray added that the WHO International Standards (IS) are primary standards and as such are produced infrequently with a substantial international collaboration required to assign values, and their usage is carefully controlled to maintain stocks and ensure continuity. Pharmacopoeial, regional, working or in-house standards are secondary standards, calibrated against the IS.
Discussion following ‘Value assignment of International Standards: challenges for potency labelling of biotechnology/biosimilar products’
There was a question about reference units, standards and dosing. Dr Gray said that, for a product such as a monoclonal antibody, which is already dosed in milligrams, there would be no attempt to change the dosing to arbitrary units. So, the activity in terms of the international unit is really for the control of the bioassay, this does not impact on the dosing and potency labelling of these products. The aim of such standards would be to aid manufacturers in monitoring consistency of production. For other biological products that do label potency in international units, such as for the coagulation factors, the continuity of the unit is ensured by calibrating against the existing standard, in order to have traceability of the unit each time a replacement standard is made. However, some slight variability is inevitable because batches of such materials are unique, and there may be some drift in potency depending on the assay and the materials themselves.
Conclusion
Colombia’s health service has improved greatly over the past 25 years and standards of heath care and access to treatments are high. Over the last seven years the country has been developing a regulatory framework to allow biosimilar products to enter the market and Colombia is now at a point where it can start implementing regulatory approval policy for biologicals/biosimilars. The educational workshop and the scientific meeting created a platform for those involved with biological/biosimilar regulation in Colombia to meet with experts from Europe and the US and a number of interactive discussions took place. The attendees shared ideas with the speakers and received clarification on issues of interest and concern.
Speaker Faculty and Moderators
Speakers
2016 Educational Workshop
Niklas Ekman, PhD, Finland
Thijs J Giezen, PharmD, MSc, PhD, The Netherlands
Gustavo Grampp, PhD, USA
Elwyn Griffiths, DSc, PhD, UK
Professor Andrea Laslop, MD, Austria
Robin Thorpe, PhD, FRCPath, UK
2017 Scientific Meeting
Johanna Andrea Garcia Cortes, MSc, Colombia
Elaine Gray, PhD, UK
Professor Tore Kristian Kvien, MD, Norway
Jennifer Liu, PhD, USA
Paul Matejtschuk, PhD, CChem, UK
Sundar Ramanan, PhD, USA
Moderators and Co-moderators
2016 Educational Workshop
Jeannette Daza Castillo
Andrey Forero Espinosa
Angélica Fula Arguello
Diego Alejandro Gutierrez Triana
Inés Elvira Ordoñez
Claudia Yaneth Niño C
Acknowledgement
The Generics and Biosimilars Initiative (GaBI) wishes to thank Dr Robin Thorpe and Professor Andrea Laslop, chair and co-chair of the 2016 workshop; and Dr Elaine Gray and Dr Paul Matejtschuk, chair and co-chair of the 2017 meeting; for their strong support through the offering of advice and information during the preparation of the workshop and meeting, respectively. Further, GaBI wishes to thank Mr Francisco Javier Sierra Esteban of INVIMA for his feedback and comments on this Meeting Report.
The authors would like to acknowledge the help of all the workshop and meeting speaker faculty and participants, each of whom contributed to the success of the workshop and meeting and the content of this report, as well as the support of the moderators and co-moderators for the 2016 workshop: Jeannette Daza Castillo, Andrey Forero Espinosa, Angélica Fula Arguello, Diego Alejandro Gutierrez Triana, Inés Elvira Ordoñez, Claudia Yaneth Niño C, in facilitating meaningful discussion during the parallel group discussions; and presenting the discussion findings at the 2016 workshop.
Lastly, the authors wish to thank Ms Alice Rolandini Jensen, GaBI Journal Editor, in preparing this meeting report manuscript and providing English editing support on the group summaries and for finalizing this manuscript.
Competing interests: The workshop and meeting were sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Elaine Gray, PhD
Principal Scientist, Haemostasis Section Biotherapeutics Group
Paul Matejtschuk, PhD, CChem
Principal Scientist – Standardisation Science
National Institute for Biological Standards and Control (NIBSC), Blanche Lane, South Mimms, Potters Bar, Hertfordshire EN6 3QG, UK
Robin Thorpe, PhD, FRCPath
Deputy Editor-in-Chief, GaBI Journal
References
1. Generics and Biosimilars Initiative. First INVIMA Educational Workshop on Assessment of Similar Biotherapeutic Products 2016; 14 June 2016; Bogotá, Colombia. Available from: www.gabi-journal.net/about-gabi/educational-workshops/first-invima-educational-workshop-on-assessment-of-similar-biotherapeutic-products-2016
2. Generics and Biosimilars Initiative. Second Colombian Scientific Meeting on Quality Assessment of Biosimilars/Similar Biotherapeutic Products 2017; 15 August 2017; Bogotá, Colombia. Available from: www.gabi-journal.net/second-colombian-scientific-meeting-on-quality-assessment-of-biosimilarssimilar-biotherapeutic-products-2017
3. Generics and Biosimilars Initiative. First Latin American educational workshop on similar biotherapeutic products; 20 January 2015; Mexico City, Mexico. Available from: www.gabi-journal.net/first-latin-american-educational-workshop-on-similar-biotherapeutic-products-mexico-city-mexico-20-january-2015.html
4. Walson PD, Thorpe R. First MENA educational workshop on regulation and approval of similar biotherapeutic products/biosimilars, Dubai, United Arab Emirates, 1 September 2015. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):173–7. doi:10.5639/gabij.2015.0404.039
5. Annese V, Avendaño-Solá C, Breedveld F, Ekman N, Giezen TJ, MSc, Gomollón F. Roundtable on biosimilars with European regulators and medical societies, Brussels, Belgium, 12 January 2016. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(2):74–83. doi:10.5639/gabij.2016.0502.019
6. Walson PD, Thorpe R. First Turkish interactive workshop on regulation and approval of similar biotherapeutic products/biosimilars, 2–3 March 2016, Ankara, Turkey. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(3):134–8. doi:10.5639/gabij.2016.0503.034
7. García Cortes JA, Sierra Esteban FJ. Regulations for biotherapeutics approval in Colombia. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(1):26-8. doi:10.5639/gabij.2018.0701.006
8. Perks B. Randomized non-inferiority trial fails to find inferiority switching from infliximab originator to CT-P13 biosimilar. Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(4):188-9. doi:10.5639/gabij.2017.0604.042
|
Author for correspondence: Robin Thorpe, PhD, FRCPath, Deputy Editor-in-Chief, GaBI Journal |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


