Roundtable on biosimilars: pharmacovigilance, traceability, immunogenicity, 15 November 2016, Madrid, Spain
Published on 2017/03/01
Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(1):31-7.
Author byline as per print journal: Professor Francisco José de Abajo, MD, MPH, PhD; Professor Joan Albanell, MD; Olga Delgado Sanchez, PharmD; Kevin Klein, MSc; José Vicente Moreno-Muelas, MD; Sol Ruiz, PhD; Professor Maria Jesús Sanz Ferrando, PhD; Robin Thorpe, PhD, FRCPath; Professor Francisco Zaragozá, PhD
|
Introduction: Biosimilars can offer a lower-cost alternative to current biological therapies and could help contribute to the much-needed savings for the healthcare systems. All biosimilars approved by the regulators must show comparable efficacy and safety with the reference biologic. However, the acceptance by the healthcare professionals and by patients is not uniform within Europe. Therefore, it is important to understand the barriers to biosimilar uptake. Increased awareness amongst the stakeholders – regulators, prescribing physicians, medical societies, pharmacists and patients – to the barriers and plausible solutions could help improve further uptake of biosimilars. |
Submitted: 2 February 2017; Revised: 25 February 2017; Accepted: 27 February 2017; Published online first: 3 March 2017
Introduction
Biosimilars offer a prospect of savings to the health systems given they are generally priced lower than the reference product. The uptake of biosimilars across Europe has not been uniform across all countries. The challenges posed by biosimilars – which are different versions of the innovator biological medicines on which they are based – are viewed in different ways. Biosimilar uptake has been relatively slow in Spain, where physicians do not appear to be readily in favour of the extrapolation of indications, despite guidance otherwise from the European Medicines Agency (EMA). There are also concerns around the effectiveness of current pharmacovigilance systems.
The Generics and Biosimilars Initiative (GaBI), with its mission to foster the worldwide efficient use of high quality and safe medicines at an affordable price, organized a roundtable discussion on biosimilars (focussing on pharmacovigilance, traceability and immunogenicity) for Spanish physicians, pharmacists and medical societies. The aim of the meeting was to learn about the challenges faced in Spain as a steadily growing number of biosimilars are approved and enter the European market.
The main themes of the meeting were on the practical challenges in the implementation of robust pharmacovigilance systems; and how to establish pharmacovigilance schemes to enhance traceability and post-marketing surveillance of biologicals including biosimilars.
The meeting was organized in collaboration with the Faculty of Medicine and Pharmacy of the University of Alcalá, and chaired by Professor Francisco Zaragozá, past-president of both the Spanish Society for Pharmacology and the European Pharmaceutical Advisory Committee. He is currently Professor of Pharmacology at the Faculty of Medicine and Pharmacy, University of Alcalá, Madrid, Spain.
Presentations were made by European speakers as well as local academic faculty, clinicians, pharmacists and Spanish regulators. The list of speakers and panel members is detailed at the end of this report, and all speakers’ presentations are available on the GaBI Journal website (www.gabi-journal.net/about-gabi/educational-workshops).
Methods
On 15 November 2016, GaBI held a Roundtable on Biosimilars in Madrid, Spain, with participation by physicians and pharmacists from Spain, as well as other experts from Europe. The programme offered speaker presentations and a discussion session to provide participants with current views on biosimilars in Spain. Presentations were in Spanish or English (with simultaneous translation).
Results
Biological medicines
The Roundtable was opened by the Chair, Francisco Zaragozá, Professor of Pharmacology at the Faculty of Medicine and Pharmacy, University of Alcalá, Madrid, Spain. Professor Zaragozá gave an overview of biological medicines including biosimilars, covering both the promise and the concerns that surround these therapies. The greatest promise of biosimilars is that they will drive down healthcare costs, both in terms of the cost of treatment and the savings made by cutting hospital readmissions. But are the concerns justified?
The introduction of therapeutic monoclonal antibodies, such as infliximab, was a notable milestone. Two biosimilars of infliximab (Inflectra and Remsima) were approved by EMA in 2013, and Inflectra was approved by the US Food and Drug Administration (FDA) in 2016.
Unlike generics which have the same active ingredient as the reference drug, the structural differences between the biosimilars and the reference biological was a concern for Professor Zaragozá. Thus, ‘We need to be very cautious in our approach’ in the use of biosimilars, he warned, calling on regulators to improve communication by providing more clarity on their rationale for the decisions.
EMA approval requirements on biosimilars: clinical aspects
Dr Sol Ruiz, Head of Biotechnology and Advanced Therapies at the Spanish Agency for Medicines and Medical Devices (Agencia Española de Medicamentos y Productos Sanitarios) and chair of EMA’s Biologics Working Party, discussed the quality and clinical aspects of EMA approval requirements for biosimilars. Dr Ruiz began by presenting a list of all biosimilars approved by EMA to date [1], demonstrating that the number of biosimilars is increasing year-over-year.
The procedure of establishing biosimilarity is a step-wise process that can be split into three categories: extensive characterization (of physicochemical structure and biological activity); in vitro functional activity (analysing all possible modes of action [MoA] of the molecule); and efficacy and safety studies (using the most sensitive or relevant population to detect differences).
Biosimilarity from a clinical perspective starts by showing a comparable pharmacokinetic (PK) profile in a sufficiently sensitive and homogeneous population of healthy volunteers, if possible. For pharmacodynamic (PD) studies, the use of multiple PD markers is recommended if possible. PD markers can be used as pivotal evidence for comparable efficacy if a clear dose-response relationship is shown, and if at least one PD marker is an accepted surrogate for efficacy, i.e. can be related to patient outcome.
Comparative studies looking at efficacy and safety must by adequately powered, randomized, preferably double blind and are normally equivalence trials. Any deviation from relevant efficacy guidelines should be justified. For these studies, usually the most sensitive patient population is preferred.
Dr Ruiz summarized the clinical studies carried out to establish the biosimilarity of CT-P13 (Remsima) with its innovator infliximab. In June 2013, CT-P13 (with the brand names Remsima [Hospira] and Inflectra [Celltrion]) were recommended for approval by EMA’s Committee for Medicinal Products for Human Use (CHMP) and is available in the European Union (EU). Inflectra was subsequently approved by FDA in April 2016, following a submission by Celltrion and its partner Pfizer.
Extrapolation of indications continues to be the most controversial issue affecting the uptake of biosimilars, said Dr Ruiz. As the list of approved biosimilars grows, regulators should make the effort to better communicate the reasoning behind the decisions made.
Evaluation of immunogenicity of biosimilars
Dr Robin Thorpe, former head of the Biotherapeutic Group at the UK’s National Institute for Biological Standards and Control, spoke about the issue of unwanted immunogenicity of biological medicines. Immunogenicity can have significant clinical consequences leading to reduced clinical efficacy, hypersensitivity or allergic reactions. Immunogenicity assessment is a vital component of regulatory submissions for product approval.
Immunogenicity assessment requires carefully planned prospective studies in a suitable indication, a well-considered strategy and a panel of appropriately validated assays for antibody detection and characterization in clinical samples. Determining the immunogenicity of biosimilar products requires similar assessment approaches to stand-alone products, but in addition comparative immunogenicity studies with the reference product are required.
Biological products can induce antibodies with different characteristics and different assays are needed to detect and measure these antibody types.
Dr Thorpe made clear that there was no reason to treat approved biosimilars any differently from all biologicals from the immunogenicity perspective. He presented data from European public assessment reports (EPARs) showing antibody frequencies for biosimilars and their reference products. Clearly neither group – biosimilar nor reference – routinely has a higher or lower antibody frequency, see Table 1.

Immunogenicity is a serious and challenging issue. It is impossible to predict when it will happen and what clinical consequences it will have. Appropriate studies are clearly needed to assess such risks. It is important to remember that immunogenicity is not new or specific to biological therapies – it exists ‘for just about everything’, said Dr Thorpe.
The factors that influence unwanted immunogenicity are shown in Box 1.
Current guidance from EMA, FDA and World Health Organization (WHO) stipulates that testing for unwanted immunogenicity is integral to product development, that animal data is not predictive of immunogenicity in humans, and that every product needs to be evaluated for immunogenicity individually and an appropriate strategy adopted based on intended clinical use. Dr Thorpe noted that EMA’s CHMP guidelines are about to be updated.
There is no perfect assay for antibody testing. CHMP recommends a tiered approach to immunogenicity testing: starting with screening assays (to identify anti-therapeutic antibodies); followed by confirmatory assays; and finally neutralization assays (to discriminate between neutralizing and non-neutralizing antibodies, see Figure 1.

Even though immunogenicity assessment strategy needs to be established on a case-by-case basis (product, patient, expected clinical parameters), unwanted immunogenicity always remains a possibility. Immunogenicity can arise throughout a product’s life cycle. In addition to this, post-approval assessment, as part of pharmacological surveillance, is key.
Understanding pharmacovigilance for biologicals and the current challenges in Spain
Francisco José de Abajo, Professor of Clinical Pharmacology at the University of Alcalá, Madrid, Spain began his presentation on the issue of pharmacovigilance in Spain by noting that pharmacovigilance for biological medicines is largely the same as for pharmaceuticals. Two differences, however, are that greater importance is placed on the possibility of immunogenicity, and on the impact of quality issues on safety.
Professor de Abajo reviewed a new chapter in the EMA guidelines on good pharmacovigilance practices (EU-GVP), entitled ‘Product or population specific considerations II: biological medicinal products’, which came into force in August 2016 [2]. The new chapter provides guidance on how to better monitor and manage the safety of biological medicines to optimize the safe and effective use of these products in Europe.
Professor de Abajo contrasted the classical view of pharmacovigilance, the reactive model, see Figure 2a with the current view, the proactive model, see Figure 2b.
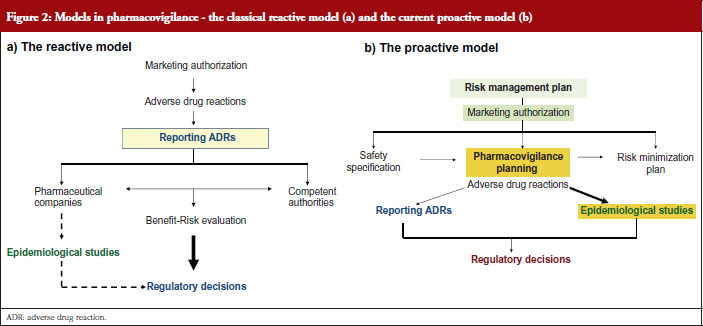
The risk management plan, see Figure 2b, aims to anticipate potential safety issues, carry out epidemiological studies, implement risk minimization measures and check the effectiveness of measures adopted.
Biologicals require particularly stringent pharmacovigilance since they are complex molecules that are considered more likely to trigger immune reactions.
Clinical data collected from patients who have been treated with biological therapies in Spain have been included in national databases in order to identify and help predict and prevent possible adverse events. The Spanish Agency for Medicines and Medical Devices’ database BIFAP (www.bifap.org), which currently includes data from eight million patients seen by general practitioners in Spain, is similar to the UK’s Clinical Practice Research Datalink (CPRD) and The Netherlands’ IPCI (Integrated Primary Care Information). Sadly, most biological treatments are dispensed in hospitals and are not included in BIFAP or in many other pharmacoepidemiological databases.
An alternative source of information is the creation of ad hoc registries of exposed patients (treated with drugs of interest) or registries of cases (patients with the disease or event of interest). An example of the former in Spain is the BIOBADASER registry – created by the Spanish Society of Rheumatology and partially funded by the Spanish Medicines Agency and several pharmaceutical companies. BIOBADASER (https://biobadaser.ser.es) provides a registry of patients treated with biologicals for rheumatic diseases. Such a registry has been successfully used to test some pharmacovigilance signals concerning these products, either confirming them, e.g. tuberculosis associated with infliximab, or refuting them, e.g. cancer associated with these biological products. This database has included in its phase II over 7,000 patients. It has helped show the effectiveness of risk minimization measures on reducing cases of tuberculosis in patients treated with infliximab. More recently, a similar initiative has been taken by the Spanish Academy of Dermatology and Venereology (AEDV) setting up a database called BIOBADADERM, which includes patients treated with biological products for dermatological conditions, e.g. psoriasis (https://biobadaderm.fundacionpielsana.es).
Professor de Abajo says the Spanish pharmacovigilance system is well prepared for the challenge of biologicals, but complementary epidemiological approaches will improve this. BIOBADASER provides a roadmap to follow.
Current challenges in traceability of biologicals – a case study from The Netherlands
Kevin Klein, a researcher at Escher, the Lygature platform for regulatory innovation in The Netherlands, spoke about a recent pilot study he has been working on looking at pharmacovigilance of biologicals.
Klein and his colleagues evaluated information-recording systems and practices in the Dutch hospital and community setting to identify determinants for brand name and batch number recording as well as success factors and bottlenecks for traceability. His team found limited traceability of brand names and batch numbers in reports of adverse drug reactions (ADRs), which they concluded might be primarily caused by the shortcomings in the recording of information in clinical practice.
To align practice with the ambition of the EU pharmacovigilance legislation regarding traceability, brand name and batch number recording needs to be (further) improved, says Mr Klein. Additional case studies in different Member States could help to map EU differences, commonalities and potential success factors for activities/interventions to improve traceability.
Considerations of the Spanish Society of Pharmacology on pharmacovigilance, traceability and immunogenicity of biosimilars from the biomedical perspective
Maria Jesus Sanz Ferrando, President of the Spanish Society of Pharmacology and Professor of pharmacology at the University of Valencia, highlighted the Spanish Society of Pharmacology’s concerns at the level of pharmacovigilance, traceability and immunogenicity of biologicals including biosimilars, specifically monoclonal antibodies.
Professor Sanz acknowledged that the introduction of biosimilars was a great advance for Spain’s national health system, but strong pharmacovigilance that includes both health professionals (including doctors, pharmacists and nurses) and patients in the communication of efficacy and adverse reactions of all products is needed.
Further development of techniques aimed at detecting the possible immunogenicity of each product is required. The Spanish Society of Pharmacology recommends that prescription of biosimilars be made by brand name, batch number and manufacturer to enable traceability in case of adverse events. The Society holds that extrapolation to other indications without previous clinical trials will require advances in in vitro studies and more sophisticated and reliable animal models.
Antibody biosimilars in oncology: an analytical to clinical perspective
Professor Joan Albanell, member and coordinator of the Spanish Society of Medical Oncology’s Biosimilars Working Group and Professor of Oncology at Pompeu Fabra University, warned against the interchangeability of biosimilars and reference drugs without specific clinical studies on the same indication in support of its safety and efficacy.
The Spanish Society of Medical Oncology’s (SEOM) position on biosimilar monoclonal antibodies is that these drugs are unlike other biosimilars (like erythropoietin and colony stimulating factors), which have easy to measure efficacy surrogate markers, e.g. haemoglobin concentrations and leukocyte levels. The challenges posed by monoclonal antibodies, such as Trastuzumab, lie in their structural complexity, their role in the treatment of a wide range of tumours, the limited correlation between efficacy surrogate markers and clinical benefits, and the heterogeneity of their mechanisms of action.
SEOM’s position on the possibility of extrapolating from a specific trial with a homogeneous population with a clinical endpoint capable of detecting differences in activity to other indications in which the drug’s mechanisms of action, the disease biology and the treatment objectives can be different, is that it should be done only on a case-by-case basis and when the mechanism of action is clear.
Since the biosimilar and the reference biological are different drugs, argues SEOM, interchangeability should not be automatic at the time of dispensing and it can only be acceptable in certain cases, with clinical justification. The situation would be different, says Professor Albanell, if there were specific clinical studies proving the safety of interchangeability at an individual level, as regulated by FDA. However, viability of these clinical trials is difficult.
Professor Albanell noted that biosimilar manufacturers have only limited knowledge of the reference product, see Figure 3, and that monoclonal antibodies are heterogenous complex proteins that are sensitive to manufacturing conditions. For example, differences in glycosylation may lead to differences in biological function, see Figure 4.


The introduction of monoclonal antibody biosimilars in oncology is welcomed given their potential to reduce treatment costs, but their multiple, not fully understood, mechanisms of action and post-translational modifications, coupled with the complexity of cancer biology, poses a significant challenge.
SEOM agrees with the need to prescribe by brand name and, further, requires that before introducing a biosimilar in a hospital, adequate circuits be established for prescription, dispensing, administration and registration using the brand name. Pharmacovigilance of biosimilars is regulated as obligatory at a European level to rule out differences with the original biological in relation to efficacy or toxicity among the healthy population.
Position statement of the Spanish Society of Rheumatology on biosimilar drugs. Clinical vision
Dr José Vicente Moreno-Muelas, former president of the Spanish Society of rheuma-tology (SER) and faculty member at the Rheumatology Department of the University Hospital Vall d’Hebron in Barcelona, Spain welcomed the arrival of biosimilars, which have opened access to biological therapy for rheumatology patients. But, echoing concerns voiced elsewhere at this Roundtable meeting, there are several areas of concern – the issues of substitution by the hospital pharmacy, automatic switching between the innovator and the biosimilar, and the need for improved pharmacovigilance.
SER holds that the decision to prescribe a biosimilar or its innovator lies solely with the prescribing physician. The exchange of an innovator for its biosimilar is a medical act that must be performed exclusively by the prescribing physician, with the consent of the patient.
Since biosimilar drugs are subject to a safety follow-up similar to that of their reference drugs, it is necessary to create specific pharmacovigilance records. SER has extensive experience in these registries and offers to carry out these security studies.
As biosimilars currently have the same international non-proprietary name (INN) as their innovators, prescription must be made by brand name in order to achieve adequate traceability. SER states that the demonstration of efficacy and safety of a biosimilar for a given indication may not be the same as for a second indication in which the reference biological drug has demonstrated efficacy and safety. When the reference drug has more than one indication, the extrapolation of indications should be justified in accordance with EMA standards. SER argues that, if necessary, each authorized indication should be individually demonstrated with double-blind, randomized, direct comparison clinical trials with the reference drug [3]. SER’s position will be updated periodically in the light of new evidence, most likely within the next two years.
Biosimilars: something more than clinical evidence
Despite the evidence of efficacy, a solid regulatory framework and the experience of switching and safety of biosimilars, the uptake of biosimilars in European markets has been patchy, notes pharmacist Dra Olga Delgado, who heads the pharmacy department at the University Hospital Son Espases, Palma de Mallorca, Spain. Dra Delgado welcomes the introduction of biosimilars, and presented evidence of the clear benefits that increased biosimilar uptake can have on reducing the cost of treatment.
She gave examples of how uptake of biosimilars varied between hospitals in the Balearic Islands and can be lower than 10% and even down to 0% in one hospital. Dra Delgado argued that healthcare professionals clearly needed more than just evidence to implement biosimilars in practice. Her approach to this challenge is that more incentives are needed for clinicians, together with more education and support for patients, and an agreement to share the savings offered by biosimilars over their innovators. Biosimilars are an opportunity for collaboration.
Conclusions of the Roundtable meeting
The opportunities offered by biosimilars were welcomed by all speakers. Realizing these opportunities will take much open discussion and sharing of information between everyone involved in the uptake of biosimilars – prescribing physicians, medical societies, patients, pharmacists and regulators.
There was broad agreement that improved pharmacovigilance is needed. The importance of recording the brand name, as well as prescribing by brand name, was noted by the Spanish Society of Pharmacology, the Spanish Society of Rheumatology and the Spanish Society of Medical Oncology. Likewise, the importance of recording the batch number was noted by the Spanish Society of Pharmacology. Both batch number and brand name recording were highlighted by Mr Kevin Klein, whose research in The Netherlands shows that improvements in recording these details are needed in order to meet EU pharmacovigilance legislation.
The role of registries – of both patients and cases – was also raised. The Spanish Society of Rheumatology has created the BIOBADASER registry with support from the pharmaceutical industry and the Spanish Medicines Agency. This registry of patients with rheumatic diseases treated with biologicals has helped support pharmacovigilance and shown the effectiveness of risk minimization measures, for example, by reducing cases of tuberculosis in patients treated with infliximab. Similarly, the Spanish Academy of Dermatology and Venereology (AEDV) is setting up a database called BIOBADADERM for patients with dermatological diseases treated with biologicals. Risk minimization is key to the risk management plan, which lies at the heart of the currently accepted proactive model of pharmacovigilance.
The risk management plan was used to allay pharmacists’ and prescribers’ fears that they were not being made aware of any changes made to the formulation of innovator or biosimilar medicines. With a risk management plan in place, such changes will be recorded appropriately, followed closely and the information made available. Some prescribers complained that they were not told when manufacturing changes are made to biologicals.
It was clear that the authorization of biosimilars by regulatory bodies, such as EMA, does not automatically persuade prescribers to stop prescribing innovator biologicals in favour of biosimilars. A similar outcome was seen at a recent GaBI Roundtable in Brussels where regulators and physicians discussed access to biological therapies [4]. More easily accessible, clear information on the reasoning behind regulatory approvals is needed. The structural differences known to exist between innovators and their biosimilars is a great concern to physicians, particularly those treating complex and not completely understood diseases like cancer. Regulators will need to improve their communication and education in this area if biosimilar uptake is to increase.
The Roundtable concluded with agreement that continued dialogue between all stakeholders will be key. The concerns raised by physicians in Spain need to be heard and addressed if biosimilar uptake is to increase.
Speaker faculty and interactive discussion session panel members
Speaker faculty
Professor Francisco José de Abajo, MD, MPH, PhD, Spain
Kevin Klein, MSc, The Netherlands
Sol Ruiz, PhD, Spain (Co-Chair)
Robin Thorpe, PhD, FRCPath, UK
Professor Francisco Zaragozá, PhD, Spain (Chair)
Interactive discussion session panel members
Professor Joan Albanell, MD, Spain
Olga Delgado Sanchez, PharmD, Spain
José Vicente Moreno-Muelas, MD, Spain
Professor Maria Jesús Sanz Ferrando, PhD, Spain
Acknowledgement
The Generics and Biosimilars Initiative (GaBI) wishes to thank Professor Francisco Zaragozá for his strong support through the offering of advice and information during the preparation of this Roundtable meeting.
The authors wish to thank Dr Beatrice Perks, GaBI Journal Editor, in preparing this meeting report and providing English editing support for finalizing this manuscript.
Competing interests: The Roundtable meeting was sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; internally peer reviewed.
Authors
Professor Francisco José de Abajo, MD, MPH, PhD
Professor Joan Albanell, MD
Olga Delgado Sanchez, PharmD
Kevin Klein, MSc
José Vicente Moreno-Muelas, MD
Sol Ruiz, PhD
Professor Maria Jesús Sanz Ferrando, PhD
Robin Thorpe, PhD, FRCPath
Professor Francisco Zaragozá, PhD
References
1. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2017 Feb 25]. www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
2. European Medicines Agency. Guideline on good pharmacovigilance practices (GVP) Product – or Population-Specific Considerations II: Biological medicinal products. EMA/168402/2014. 4 August 2016 [homepage on the Internet]. [cited 2017 Feb 25]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/08/WC500211728.pdf
3. Abad Hernández MA, Andreu JL, Caracuel Ruiz MA. Position paper from the Spanish Society of Rheumatology on biosimilar drugs. Reumatol Clin. 2015;11(5):269-78.
4. Annese V, Avendaño-Solá C, Breedveld F, Ekman N, Giezen TJ, Gomollón F, et al. Roundtable on biosimilars with European regulators and medical v, Brussels, Belgium, 12 January 2016. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(2):74-83. doi:10.5639/gabij.2016.0502.019
|
Author for correspondence: Robin Thorpe, PhD, FRCPath, Deputy Editor-in-Chief, GaBI Journal |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2017 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


