Tyrosine kinase inhibitors becoming generic drugs – risks and chances from a regulatory perspective
Published on 2014/03/24
Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(2):79-87.
Author byline as per print journal: Niels Eckstein, PhD; Lea Röper, BSc; Bodo Haas, PhD; Henrike Potthast, PhD; Ulrike Hermes, PhD; Christoph Unkrig, MD; Frauke Naumann-Winter, PhD; Harald Enzmann, MD
|
Aim: To provide a systematic overview on: i) safety profiles; ii) pharmacokinetic parameters; and iii) regulatory framework of anti-cancer tyrosine kinase inhibitors (TKI). |
Submitted: 7 February 2014; Revised: 13 March 2014; Accepted: 13 March 2014; Published online first: 26 March 2014
Introduction
Initially, great expectations were associated with these drugs; some were met, others not. Tyrosine kinase inhibitors (TKIs) are a very worthy additional option for physicians in clinical management of certain types and lines of treatment of cancer, see Table 1 for a tabular overview. In haemato-oncology, they are contributing to the tendency of chronificating rather than curing the disease. In contrast, the expectation of a new era of cancer-therapy without or at least substantially less side effects were not fulfilled, TKI have numerous, partly severe side effects eventually entailed with fatal outcome, see Table 2. On the other hand, after evolving of resistance to conventional (cytotoxic) or targeted anti-cancer therapy, TKI serve as additional therapy options in second, third and/or fourth-line therapy regimes according to their approved indications. For instance, Sunitinib is approved after Imatinib resistance formation in gastrointestinal stromal tumours (GIST), and Lapatinib after non-responding to antracycline- or taxane-based chemotherapy in combination with Trastuzumab in HER-2 positive breast cancer. Taken together, TKI are a valuable extension of the cancer drug armamentarium [1, 2].


Challenges of generic TKI drugs in cancer therapy
According to their European birth date during the past decade, these substances successively will be running off-patent within the next years, see Table 1. From a regulatory point of view, this raises the question how marketing authorization applications (MAA) should be filed and especially, how therapeutic equivalence should be established for generic applications. In general, demonstrated bioequivalence (BE) allows generic medicinal products to refer to the efficacy and safety data of the originator medicinal product. It is easy to anticipate, that numerous questions in this regard will arise in the near future.
Aqueous (non-complicated) intravenously applied drug products have a 100% bioavailability directly per definition, thus, no BE studies are required for a MAA of such generic drugs. However, for orally applied drug products, BE with the originator product needs to be shown, which may be done using patients or healthy volunteers in respective in vivo studies or by means of comparative in vitro investigations.
Since decades BE-acceptance criteria for area under the curve (AUC) and, maximum plasma concentration (Cmax) require the 90% confidence intervals being completely within 80–125% (for AUC and Cmax) to assume BE. The acceptance range may be tightened to 90–111% for one or both pharmacokinetic (PK) characteristics according to the European BE-Guideline [3] in the case of narrow therapeutic index drugs (NTID). In cases of class I and III compounds having identified not to have a narrow therapeutic index – specific in vitro dissolution data may substitute for human BE-studies considering also particular requirements on excipients. This concept follows the principles of the biopharmaceutical classification system (BCS) [3].
It is likely that numerous questions in regard to the appropriate data package will arise in the near future including questions on the appropriate study design, on the appropriate study population, nutrition status, single or repeated dose design, appropriate BCS classification of the individual compound or the classification as NTID.
MAA for new generics may be processed via different regulatory authorizations routes, i.e. national procedures in European Member States, decentralized procedures involving several European Member States or centralized procedures for all European Member States. As the latter is an option only for generics for which the originator medicinal products already obtained marketing authorization from a centralized procedure, this option may receive more attention with the increasing number of medicinal products with centralized authorizations that are running off data protection and patent in the next years.
With the intent to enable a consistent approach for these different routes the European Medicines Agency (EMA) issued an initiative to harmonize the data requirements throughout European Member States, i.e. EMA initiated a pro-active programme ‘Product-Specific Bioequivalence – Guidance for Generics’ [4]. EMA defines the objective of this initiative as follows: ‘Product specific guidance for the bioequivalence assessment of immediate release generic formulations should a priori be defined.’ Thus, applicants should be given a clear scientific guidance, how to design BE-studies and, thus, how to file generic applications. This programme includes BCS classifications for drug substances, so that a harmonized view on the BCS classification and consequently the appropriateness of a BCS-based biowaiver approach can be expected for respective products. Furthermore, the guidance provides information on the type of expected data, e.g. appropriate study population (patients or healthy volunteers), mode of administration (fasten or fed), single dose or steady-state design, appropriate dose strength and analytes, the classification as NTID. The first wave of 16 medicinal products is dominated by anti-infectives and TKI. Dasatinib, Erlotinib, Imatinib, Sorafenib and Sunitinib are covered in this first round of harmonization [4].
From a clinician’s point of view regarding drug safety, see Table 2, one could be tempted to assume that all anti-cancer medicinal products including TKI are considered as NTID. However, this is not the case. Different definitions of NTID by different regulatory agencies do exist. US Food and Drug Administration classification of narrow therapeutic ratio:
- less than a 2-fold difference in median lethal dose (LD50) and median effective dose values (ED50), or
- less than 2-fold difference in the minimum toxic concentrations (MTC) and minimum effective concentrations (MEC) in the blood, or
- safe and effective use of the drug products requires careful titration and patient monitoring.
In contrast to the US, for the EU no list of substances with NTID-designation is available. So far the consideration of a given substance as NTID is mainly based on national traditions. Only for a few medicinal substances, e.g. Ciclosporine, Tacrolimus, a harmonized EU decision was issued by a referral procedure. According to the draft ‘Product-Specific Bioequivalence – Guidance for Generics’ no drug is newly considered as NTID, only Tacrolimus is considered as such based on the previously finalized referral procedure.
According to the European Bioequivalence Guideline [3] clinical considerations are the basis for NTID decisions. Thus, safety and efficacy profile have to be taken into account.
Most conventional cytotoxic medicinal products are given parenterally for a short duration in repeated cycles. They are mostly dosed on an individual basis, e.g. body surface or weight. The recommended dose is normally the maximum tolerated dose (MTD) or close to it.
Marketed TKI drugs are typically given continuously via the oral route and at a flat dose. Although a most effective and durable target saturation is the primary objective for dose development of TKI drugs, it is obvious that for several TKI drugs the recommended dose is the same as the reported MTD, e.g. Bosutinib, Pazopanib, Ponatinib or Sunitinib, see Table 3. The dose-limiting toxicities include grade 3 gastrointestinal and hepatic toxicities, grade 3 skin toxicities, grade 3 fatigue, and grade 3 hypertension. For Sunitinib grade 2 bullous skin toxicity, grade 3 fatigue, and grade 3 hypertension are reported as dose-limiting toxicities. Furthermore, at approximately twice the therapeutic concentration a grade 2 QT-prolongation is expected (Summary of Product Characteristics Sutent [5]).
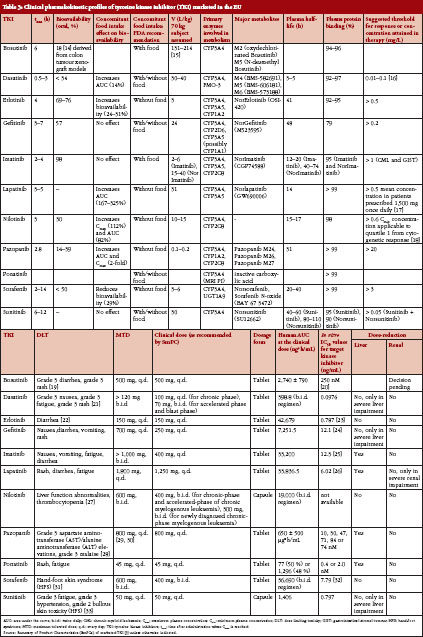
From a clinical point of view there are arguments for consideration as an NTID for selective TKI which are elucidated for the example of Sunitinib: The dose of 50 mg/d is the recommended dose for renal cell carcinoma and the MTD at the same time. The documented adverse events (AE) and adverse drug reactions (ADR) are serious, and toxicity may be difficult to control due to long half-life of parent compound and main metabolite (40–60 h and 80–110 h, respectively). The described toxicity induces a high probability of dose reductions with the intent to reduce exposure. The patient safety may be impaired in case of an exchange between originator and generic medicinal product following dose reduction: dose reductions of 12.5 mg represent a 25% and 33% decrease from the recommended dose for renal cell carcinoma and neuroendocrine tumours of pancreatic origin, respectively. In case of exchange of the originator for a generic drug the AUC from the reduced dose of the generic may be nearly the same as the AUC from the normal dose of the originator if normal acceptance criteria for BE (90% CI for AUC and Cmax 80–125%) are applied.
From a safety point of view it should be mentioned that chronic exposure to a dose that was identified as the maximum tolerable dose in a short-term study may render the tolerable short-term toxicity into intolerable long-term toxicity.
Safety of certain TKI
Dasatinib, Nilotinib and Bosutinib – CML (chronic myeloid leukaemia) – TKI with different safety profiles from a regulatory point of view and availability of second generation TKI
In general TKI are well tolerated in clinical practice, particularly, if compared with the toxicity of cytostatic drugs normally used in oncology. Often side effects are only mild (grade 2 and lower) and occur early in the treatment course. Frequently they last only some days or weeks and resolve spontaneously. Moreover, even if drug-related toxicity requires drug discontinuation, re-exposition is often successful and permanent dose reduction is rarely necessary.
The advent of Imatinib in 2001 has dramatically changed the prognosis in patients with chronic myeloid leukaemia (CML): The five-year survival rate of patients with chronic phase CML improved from approximately 20% in the pre-TKI era to more than 90% [6]. In those patients who achieve a stable cytogenetic response with Imatinib overall survival is reported with 95.2% at 8 years in the literature and thus does not differ statistically significantly from that of the general population [7]. Imatinib is still the most common TKI modality used as a front-line therapy in CML across the world. However, due to the occurrence of Imatinib resistance and intolerance, second generation TKI as Dasatinib, Nilotinib and Bosutinib have been developed. In non-clinical models they are 30 to 300 times more potent than Imatinib and can inhibit most Imatinib-resistant BCR-ABL mutations (EPARs for Imatinib, Dasatinib and Nilotinib [4]). Comparable with the experience in anti-infective drugs, multidrug-resistant BCR/ABL mutations occurs which preclude further use of the approved TKI. For example, patients with T315I mutation respond only on treatment with third generation TKI Ponatinib, which was specifically designed as a treatment option for these populations.
TKI indicated in CML have some side effects in common as myelosuppression, gastrointestinal complaints, rash, fatigue, headache and peripheral and periorbital oedema; however, intensity varies significantly between the different products. Other AE are peculiar of each drug: Imatinib has been uncommonly associated with severe heart failure, while Nilotinib is associated with QT prolongation, pancreatitis, increased rate of cardiovascular events, and occurrence of peripheral arterial occlusive disease (PAOD). Dasatinib may cause pleural, pericardial and peritoneal effusions; additionally interaction with platelet function is discussed to explain higher rates of gastrointestinal bleeding observed in clinical practice. Bosutinib is associated with significant gastrointestinal toxicity (diarrhea) and hepatotoxicity. Serious AE observed with Ponatinib are an alarming high rate of arterial thrombosis, and cardiovascular events as well as hepatotoxicity.
Differences in the safety profiles of these TKI seems to be at least partially explained by the additional inhibition of other signalling pathways apart BCR-ABL [c-Kit, Src family kinases, platelet-derived growth factor receptors (PDGFR), and others].
However, it should be kept in mind that TKI treatment of CML has to be administered lifelong and knowledge about potential long-term risks and efficacy, especially for the second generation TKI Dasatinib, Nilotinib and Bosutinib, is still limited. Whether risks associated with Ponatinib treatment can be tolerated is currently under discussion again.
Not only from a regulatory perspective careful attention on recommended risk minimization measures as defined in the product information is at the end essential to avoid treatment complications that may completely jeopardize the sought treatment success.
Can TKI be curative in CML? – Current strategies to avoid emergence of resistance
The availability of at least five TKI approved for the treatment for CML and the emergence of drug resistance and intolerance have induced a lively and complex debate on the best strategy to optimize TKI treatment.
Currently, CML treatment is ruled by the paradigm that patients with newly diagnosed CML who respond rapidly to initial treatment with a TKI and who do not develop severe intolerance against TKI should continue TKI therapy lifelong. On the other hand, long-term follow up of patients treated in the pivotal trials for Imatinib, Dasatinib and Nilotinib has shown that a relevant proportion of these patients not only achieved complete cytogenetic response but also sustaining reductions in BCR-ABL transcripts below the lower level of quantification (‘complete molecular response’). Stability of BCR-ABL absence over years in these patients has led to speculations that these patients are cured of CML.
However, can treatment be discontinued safely in patients without evidence for minimal residual disease? Results from two studies (STIM; TWISTER) [8, 9] indicate that if TKI (Imatinib) is discontinued, more than half are expected to relapse within one year. At two years post-discontinuation, 47% of patients in TWISTER were still relapse free and remained off therapy. Those with molecular relapse responded to re-treatment, but patients may not attain the same degree of response upon re-treatment, which illustrates the potential risks of treatment discontinuation approach. As data available is derived from relatively small studies and follow-up is limited, larger studies are needed (and planned) to determine further the safety of such strategies. Thus, current CML treatment guidelines still recommend lifelong therapy with TKI.
Other open topics in the scientific discussions on TKI treatment in CML reflect the question, whether a more rapid and deeper molecular response observed for second generation TKI like Dasatinib and Nilotinib is clinically relevant and indicates a better prognosis. In this context high dose treatment (‘MTD approach’) or sequential therapy with different TKI to lower development of resistance are under discussion.
The outstanding progress made in this area of therapy is best illustrated by the fact that since approval of Imatinib the ‘gold standard’ endpoint ‘overall survival’ is no longer sufficiently discriminative for clinical trials in patients with CML under TKI; surrogate marker as ‘complete cytogenetic response’ or ‘major molecular response’ have been validated and are now accepted as efficacy correlate by regulatory agencies.
Orphan drug status of TKI
The orphan regulation aims at fostering drug development for serious or life-threatening diseases with a prevalence of less than five in 10,000 people in the EU. A sponsor may apply for orphan designation any time prior to an application for marketing authorization (usually even before clinical development). The orphan drug status then needs to be confirmed during the marketing authorization procedure. The most important incentive of the regulation is 10-year market exclusivity for an orphan medicinal product with respect to similar medicinal products. Neither EMA nor EU Member States can authorize a product, which is regarded similar with respect to chemical structure and mode of action and therapeutic indication. Generics, by definition, fulfil all of these criteria.
Imatinib is the paradigm of targeted therapy with its target, the Philadelphia chromosome, occurring in two rare forms of cancer, CML and acute lymphatic leukaemia (ALL), which remain rare in spite of recent advances for treatment. Other cancers, e.g. renal cell carcinoma, was recently reported to exceed the prevalence threshold of five in 10,000 people so that no further orphan designations are expected.
Orphan similarity and market exclusivity
In addition to the incentive of the above-mentioned 10-year market exclusivity intended by the European orphan regulation [10], there maybe a probably unintended additional incentive. Special circumstances are conceivable under which the market exclusivity granted for orphan products may exclude marketing authorization of a generic product. These special circumstances first occurred when the orphan drug Tasigna (Nilotinib) was assessed as ‘similar’ to Glivec (Imatinib). Glivec was first authorized in the EU in 2003. The Committee for Medicinal Products for Human Use (CHMP) gave a positive opinion on its benefit-risk balance, the Committee for Orphan Medicinal Products (COMP) confirmed the significant benefit and so Glivec got the most important incentive for the development of medicines for orphan diseases – the market exclusivity. Under the condition of the European orphan drug regulation no medicinal product ‘similar’ to Glivec would get marketing authorization for ten years – unless the similar product had superior efficacy or safety or the market authorization holder (MAH) of the protected product gives consent to the marketing of the similar product.
Several years after marketing authorization of Glivec was granted, similarity assessment of Tasigna concluded that Tasigna was a similar product to Glivec and the market exclusivity of Glivec would therefore be prohibitive for the authorization of Tasigna. In the context of a similarity assessment, three characteristics of a given drug are decisive:
- the chemical structure (respectively structural similarity to the innovator product)
- the molecular mechanism of action, and
- the indication.
In the first step of Tasigna marketing authorization, this was not problematic, because Tasigna was first authorized in second line after first-line therapy with Glivec. However, with the extension of indications to first-line treatment of CML, Tasigna was authorized only with the consent of the MAH of Glivec (not surprisingly, as both medicines are products of Novartis). The COMP confirmed a significant benefit and thus Tasigna received its ten own year market exclusivity beginning with the commission decision in 2007.
When data protection and orphan market exclusivity expired for Glivec generic Imatinib products to the reference product Glivec were submitted. There was, however, the previous regulatory decision that Glivec and Tasigna are similar products – including the assessment of Imatinib and Nilotinib as similar active substances based on their chemical structure and pharmacological mechanism. An authorization of a generic Imatinib product to the reference product Glivec would therefore not be granted if it violated the 10-year market exclusivity of Tasigna, which began in 2007.
It is safe to assume that the European orphan legislation was never meant to preclude the authorization of generics after the data protection and the ten years orphan protection of the reference product had expired. And it also seems that this was not a deliberate abuse of a complicated legal and regulatory situation by Novartis but rather unintended. If that had been a wicked, albeit brilliant, marketing-driven strategy, the exact alignment of the indications of Glivec and Tasigna would have effectively prevented any Imatinib generics for many years. As the indications of Tasigna and Glivec overlap for the majority of patients but are not identical, a marketing authorization for Imatinib generics restricted to the indications not granted for Tasigna became possible. This is why the indications of generic Imatinib products are different from the indications of the reference product Glivec.
Is the approved dose recommendation always the right choice – notions on Erlotinib?
Sometimes the question is raised, whether dose-regimes for pivotal phase III studies (derived from early phases of clinical development) are the right ones. Especially with Erlotinib, where different dose regimes are recommended for different tumour types and toxicity is high, the question is raised: is there a minimal effective dose (MED) in epidermal growth factor receptor (EGFR)-mutated tumours below recommended dosage (or do patients indeed have to be titrated until MTD is reached)? In this regard, dose limiting toxicities (DLT) do not have to be the same for different tumour types or even in one tumour entity for all patients, see Table 3.
Regarding Erlotinib (apart from case reports) currently one publication is available, trying to bridge in vitro evidence directly with patient data [11]. This article is pointing to the fact that the MED of Erlotinib might in fact be below the recommended dose of 100 mg (pancreatic carcinoma) and 150 mg non-small cell lung cancer (NSCLC), respectively. The rationale of this study is: ‘The usual clinical dose of Gefitinib (250 mg/day) is only one third of its MTD, while the dose of Erlotinib (150 mg/day) is at its MTD. In NSCLC cell lines both TKI have similar micromolar (μM) inhibitory concentrations.’ Therefore, the authors investigated whether Erlotinib when administered at only 25 mg/day can inhibit the mutated form of the EGFR in NSCLC. Apart from similar in vitro-IC50-values, blood concentrations of both drugs differ remarkably (by a factor of more than 5):
Csteady state (Gefitinib at 250 mg/day) = 0.5 μM
Csteady state (Erlotinib at 150 mg/day) ≥ 2.5 μM
These values were derived from early phase clinical development programmes of the respective drugs [12, 13].
The authors conclude: ‘In NSCLC cell lines, Gefitinib and Erlotinib have similar inhibitory profiles. In patients with NSCLC and EGFR activating mutations, a dose of Erlotinib 25 mg/day (equivalent to Gefitinib 250 mg/day) leads to impressive response rates and progression free survival similar to the growing experience with the approved doses of Gefitinib (250 mg/day) and Erlotinib (150 mg/day). Identifying prospectively the lowest, clinically active dose ranges of Erlotinib and Gefitinib will help further personalize care for patients with tumours harbouring EGFR mutations.’ Unfortunately, this study is limited in evidence: it is basically an in vitro study correlated with data from only seven patients. Thus, no answer on what is the MED of Erlotinib can be given by now.
Conclusion
A decade ago, TKI were introduced into clinical anti-cancer therapy. At first sight, the molecular mechanism of action appears to comprise only a targeted approach in blocking tyrosine kinases. However, this should not be misleading; numerous closely interconnected signalling pathways are involved and the complexity of TKI molecular mechanism is far from being understood completely. For clinicians, TKI are a worthy new modality of tumour-therapy amending classical cytotoxic regimes. TKI are of substantial benefit in terms of efficacy with a tolerable safety profile. However, long-term safety issues might not be fully elucidated at present and, thus, cannot be finally judged upon. Throughout the next years, many of these substances will run off-patent. Thus, regulatory guidance will be required for instance on whether certain substances like Sunitinib fulfil the criteria of a narrow therapeutic index drug. Apart from that, most TKI are orally administered, thereby raising the question whether BCS-based biowaiver can apply. In addition, design and requirements of BE-studies will be an issue in the EMA-initiative of product specific guidance on anti-cancer TKI.
Acknowledgement
The support of Mr Andreas Duda is gratefully acknowledged. This review article was supported by intramural funding of the Federal Institute for Drugs and Medical Devices (Bundesinstitut für Arzneimittel und Medizinprodukte, BfArM).
Authors’ contributions
All authors filed the manuscript, Dr Niels Eckstein and Ms Lea Röper performed a systematic search on clinical PK-parameter.
Disclaimer
The opinions mentioned throughout the manuscript are personal views of the authors and do not reflect an official position of the Federal Institute of Drugs and Medical Devices or an EMA-committee or working party, respectively.
Competing interests: The authors declare that they have no competing interests.
Provenance and peer review: Not commissioned; externally peer reviewed.
Co-authors
Lea Röper, BSc
Bodo Haas, PhD
Henrike Potthast, PhD
Ulrike Hermes, MD
Christoph Unkrig, MD
Frauke Naumann-Winter, PhD
Harald Enzmann, MD
References
1. Laurie SA, Goss GD. Role of epidermal growth factor receptor inhibitors in epidermal growth factor receptor wild-type non-small-cell lung cancer. J Clin Oncol. 2013;31(8):1061-9.
2. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341-54.
3. European Medicines Agency. Guideline on the investigation of bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1/ Corr **. January 2010 [homepage on the Internet]. 2010 Mar 10 [cited 2014 Mar 13]. Available from: http://www.ema.europa.eudocs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
4. European Medicines Agency [homepage on the Internet]. 2014 [cited 2014 Mar 13]. Available from: http://www.ema.europa.eu/ema
5. Heads of Medicines Agencies. Human MRIndex [homepage on the Internet]. 2014 [cited 2014 Mar 13]. Available from: http://mri.medagencies.org/Human/
6. Quintas-Cardama A, Cortes JE. Chronic myeloid leukemia: diagnosis and treatment. Mayo Clin Proc. 2006;81(7):973-88.
7. Gambacorti-Passerini C, Antolini L, Mahon FX, Guilhot F, Deininger M, Fava C, et al. Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib. J Natl Cancer Inst. 2011;103(7):553-61.
8. Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029-35.
9. Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Yeung DT, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood. 2013;122(4):515-22.
10. Regulation (EC) No 141/2000 of the European Parliament and of the Council on orphan medicinal products. 22 January 2000 [homepage on the Internet]. 2000 Jan [cited 2014 Mar 13]. Available from: http://eur-lex.europa.eu
11. Yeo WL, Riely GJ, Yeap BY, Lau MW, Warner JL, Bodio K, et al. Erlotinib at a dose of 25 mg daily for non-small cell lung cancers with EGFR mutations. J Thorac Oncol. 2010;5(7):1048-53.
12. Baselga J, Rischin D, Ranson M, Calvert H, Raymond E, Kieback DG, et al. Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J Clin Oncol. 2002;20(21):4292-302.
13. Hidalgo M, Siu LL, Nemunaitis J, Rizzo J, Hammond LA, Takimoto C, et al. Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J Clin Oncol. 2001; 19(13):3267-79.
14. Golas JM, Lucas J, Etienne C, Golas J, Discafani C, Sridharan L, et al. SKI-606, a Src/Abl inhibitor with in vivo activity in colon tumor xenograft models. Cancer Res. 2005;65(12):5358-64.
15. Abbas R, Hug BA, Leister C, Gaaloul ME, Chalon S, Sonnichsen D. A phase I ascending single-dose study of the safety, tolerability, and pharmacokinetics of bosutinib (SKI-606) in healthy adult subjects. Cancer Chemother Pharmacol. 2012; 69(1):221-7.
16. Birch M, Morgan PE, Handley S, Ho A, Ireland R, Flanagan RJ. Simple methodology for the therapeutic drug monitoring of the tyrosine kinase inhibitors dasatinib and imatinib. Biomed Chromatogr. 2013;27(3):335-42.
17. Burris HA, 3rd, Taylor CW, Jones SF, Koch KM, Versola MJ, Arya N, et al. A phase I and pharmacokinetic study of oral lapatinib administered once or twice daily in patients with solid malignancies. Clin Cancer Res. 2009;15(21):6702-8.
18. Giles FJ, Yin OQ, Sallas WM, le Coutre PD, Woodman RC, Ottmann OG, et al. Nilotinib population pharmacokinetics and exposure-response analysis in patients with imatinib-resistant or -intolerant chronic myeloid leukemia. Eur J Clin Pharmacol. 2013;69(4):813-23.
19. Daud AI, Krishnamurthi SS, Saleh MN, Gitlitz BJ, Borad MJ, Gold PJ, et al. Phase I study of bosutinib, a src/abl tyrosine kinase inhibitor, administered to patients with advanced solid tumors. Clin Cancer Res. 2012;18(4):1092-100.
20. Vultur A, Buettner R, Kowolik C, Liang W, Smith D, Boschelli F, Jove R. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol Cancer Ther. 2008;7(5):1185-94.
21. Demetri GD, Lo RP, MacPherson IR, Wang D, Morgan JA, Brunton VG, et al. Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin Cancer Res. 2009;15(19):6232-40.
22. Bonomi P. Erlotinib: a new therapeutic approach for non-small cell lung cancer. Expert Opin Investig Drugs. 2003;12(8):1395-401.
23. Moyer JD, Barbacci EG, Iwata KK, Arnold L, Boman B, Cunningham A, et al. Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res. 1997;57(21):4838-48.
24. Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62(20):5749-54.
25. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561-6.
26. Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1(2):85-94.
27. Kantarjian H, le CP, Cortes J, Pinilla-Ibarz J, Nagler A, Hochhaus A, et al. Phase 1 study of INNO-406, a dual Abl/Lyn kinase inhibitor, in Philadelphia chromosome-positive leukemias after imatinib resistance or intolerance. Cancer. 2010; 116(11):2665-72.
28. Yau T, Chen PJ, Chan P, Curtis CM, Murphy PS, Suttle AB, et al. Phase I dose-finding study of pazopanib in hepatocellular carcinoma: evaluation of early efficacy, pharmacokinetics, and pharmacodynamics. Clin Cancer Res. 2011;17(21):6914-23.
29. Shibata SI, Chung V, Synold TW, Longmate JA, Suttle AB, Ottesen LH, et al. Phase I study of pazopanib in patients with advanced solid tumors and hepatic dysfunction: a National Cancer Institute Organ Dysfunction Working Group study. Clin Cancer Res. 2013;19(13):3631-9.
30. Infante JR, Novello S, Ma WW, Dy GK, Bendell JC, Huff A, et al. Phase Ib trial of the oral angiogenesis inhibitor pazopanib administered concurrently with pemetrexed in patients with advanced solid tumors. Invest New Drugs. 2013;31(4):927-36.
31. Moore M, Hirte HW, Siu L, Oza A, Hotte SJ, Petrenciuc O, et al. Phase I study to determine the safety and pharmacokinetics of the novel Raf kinase and VEGFR inhibitor BAY 43–9006, administered for 28 days on/7 days off in patients with advanced, refractory solid tumors. Ann Oncol. 2005;16(10):1688-94.
32. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099-109.
33. Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol. 2006;24(1):25-35.
|
Author for correspondence: Niels Eckstein, PhD, Federal Institute of Drugs and Medical Devices, 3 Kurt-Georg-Kiesinger-Allee, DE-53175 Bonn, Germany |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2014 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



Rod, I am forwarding you this recent article published in Generics and Biosimilars Initiative Journal (GaBI Journal) on generics TKIs for your information. In particular the section that mentioned; “Aqueous (non-complicated) intravenously applied drug products have a 100% bioavailability directly per definition, thus, no BE studies are required for a MAA of such generic drugs. However, for orally applied drug products, BE with the originator product needs to be shown, which may be done using patients or healthy volunteers in respective in vivo studies or by means of comparative in vitro investigations”.