Supporting biosimilarity and extrapolation
Published on 2015/12/11
Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):180-3.
|
Abstract: |
Submitted: 28 July 2015; Revised: 15 August 2015; Accepted: 16 August 2015; Published online first: 29 August 2015
The importance of analytical and clinical data in establishing and confirming biosimilarity is reviewed, highlighting the strengths and limitations of each. While clinical studies cannot be used to determine biosimilarity, they provide important confirmation of biosimilarity. Establishing and confirming biosimilarity are central to the extrapolation of indications for biosimilars.
The European Medicines Agency (EMA) has stated that ‘if clinical similarity can be shown in a key indication, extrapolation of efficacy and safety data to other indication(s) of the reference product may be possible’ under certain conditions [1].
Clinical studies play a role in supporting biosimilarity, analytics are an essential tool for establishing biosimilarity. Analytics are typically more sensitive than traditional clinical endpoints in this respect.
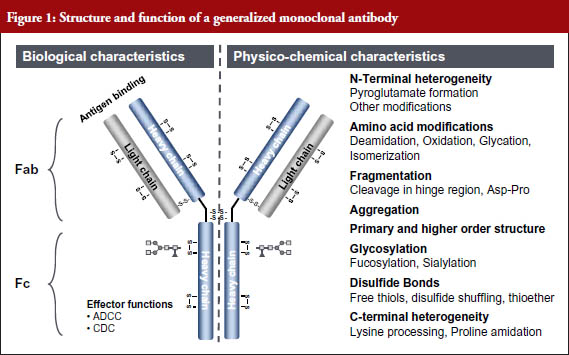
The sensitivity of analytics has increased exponentially in recent years – from a detection limit of 100 pmol in 1990 to a detection limit of 0.00001 pmol in 2011 (A 10 million-fold increase in 21 years) [2]. Because of this, all relevant structural components and functions of even a monoclonal antibody (mAb) can now be determined with high sensitivity, and the structure–function relationships determined, see Figure 1 [3].
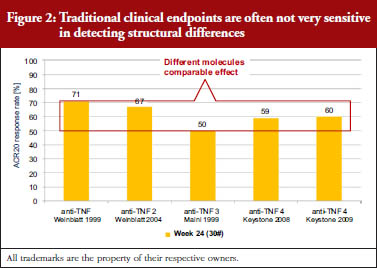
Traditional clinical endpoints are generally not very sensitive in detecting structural differences between clinical agents. In trials of arthritis treatments, for example, the effectiveness of a treatment is recorded according to American College of Rheumatology (ACR) criteria. ACR criteria include tender or swollen joint counts, patient or doctor assessments and related indicators of disease severity. ACR20 indicates a 20% improvement in ACR criteria. A review of data collected between 1999 and 2009 showed that five different anti-inflammatory anti-tumour necrosis factor (TNF) treatments shared similar ACR20 response rates, despite the fact that these were five different molecules, see Figure 2 [3]. In other words, the results of clinical trials cannot be used on their own to determine biosimilarity.
Analytics inform biosimilarity and extrapolation
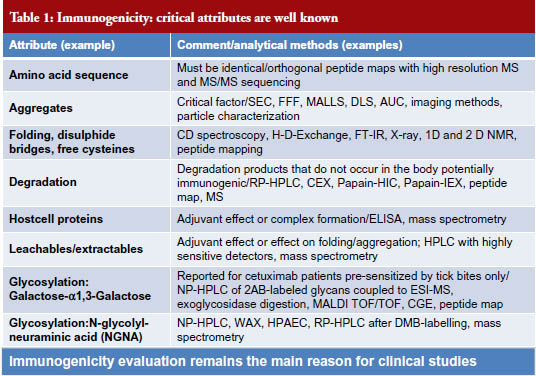
The potential clinical impact of all quality attributes of every drug must be evaluated in order to establish biosimilarity. Clinical impact includes the following components: (1) immunogenicity, which remains the main reason of clinical studies, see Table 1; (2) safety/toxicity; (3) pharmacokinetics; and (4) efficacy, see Figure 3 [3].

In the case of toxicity, it is important to consider both pharmacological toxicity, which is caused by biological activities of the molecule that have been designed to match the reference product, and off-target toxicity (understood here as caused by binding to other molecules than the target), which is rare with biologicals since they are highly specific to their target.
Pharmacokinetics looks at the attributes critical to absorption, distribution, metabolism and excretion of a drug. A list of attributes and analytical methods is shown in Table 2 [3].

There are several sources of information to draw on when considering the impact of product attributes on safety and efficacy. First, there is variability within the reference product itself, from batch-to-batch or because of changes in manufacturing. Second, the scientific literature provides observations made in clinical studies and clinical practice. The literature also contains immunological studies and functional in vitro and in vivo studies. A third source of information is found in systematic and structure–function studies – the investigation of different combinations of attributes during development. Finally, animal studies provide information on pharmacokinetics (PK), pharmacodynamics (PD) and sometimes also efficacy, if adequate disease models are available.
The criticality of each attribute must be assessed separately for each indication based on known information. The biological activities involved in each indication must be considered – the impact on PD, efficacy and safety. Likewise, the impact on PK in, and the immunological state of, different patient populations must be considered, including disease status, comorbidities and co-medication. It is important to apply a systematic approach supported by mathematical tools; additive or cancelling effects of different attributes must be taken into account. Cell-based and animal studies can provide additional information.
Sensitive indication
Regulators request the use of ‘sensitive indication’, particularly in view of extrapolation. The US Food and Drug Administration (FDA) recommends endpoints and study populations that are sensitive in detecting clinically meaningful differences in safety and effectiveness [4]. EMA recommends that the study population should be sensitive for detecting potential differences between the biosimilar and the reference product [5].
The three main points to consider when selecting a study population are immunocompetence, large effect size and feasibility. Under immunocompetence, the population must be as likely, or more likely, to show detectable differences in immunogenicity compared with other on label populations. The advantage of a population with a large effect size is that small differences are more easily detected compared with populations with a small effect size (10% of 100 is 10, which is easier to detect than 10% of 10, which is 1).
The similarity exercise
It is important to remember that extrapolation is from the reference product to the biosimilar and not from one clinically studied indication to the others, see Figure 4 [3], explained by Windisch [3]. All indications will have been thoroughly studied clinically with the reference product.
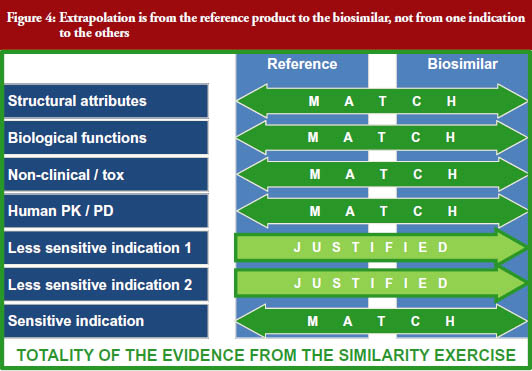
The biosimilar is connected with this wealth of data via multiple very robust bridges: (i) analytical (structural) similarity; (ii) functional similarity (biological and functional assays); (iii) human PK/PD similarity; and (iv) similarity in a clinical study in an indication which is sensitive in detecting differences in immunogenicity and efficacy.
For extrapolation, the structural elements relevant to immunogenicity and to the mechanism(s) of action in the different indications are especially important.
If there is a difference in a potentially functionally relevant attribute, it must be evaluated if this difference could have clinical consequences. This can be done in two ways. First, choose an indication for the clinical safety/efficacy study which has a mechanism of action in which this function is involved. Second, provide data from cell-based assays or in vivo non-clinical studies which prove that the difference is not relevant. The best situation is to avoid differences in potentially relevant functional attributes altogether.
US-based injectables specialist Hospira, a global leader in biosimilars, has called on regulators to continue to adopt important scientific and clinical concepts of extrapolation. Europe has 21 EMA-approved biosimilars and FDA approved its first biosimilar in the US in 2015. Hospira released a white paper in April 2015 calling for continued support for extrapolation in order to drive increased patient access to important biological medicines [6]. Hospira depicts a somewhat different scientific approach to extrapolation than that of Windisch [3]. The company notes that the introduction of biosimilar filgrastim in the UK, by Hospira and other companies, had increased patient access by 50 per cent.
Infliximab extrapolation
Taking a higher focus on extrapolation than was covered in the Windish presentation [3] reviewed above, it is clear that the principle of extrapolating indications for biosimilars, and the science involved, are not well understood by all physicians [7]. This can lead to distrust of biosimilars, and low uptake. Members of EMA’s Biosimilar Medicinal Products Working Party (BMWP) addressed these concerns using extrapolation of indications in several biosimilars, including infliximab, as examples [8]. The working party analysed data published in this area and concluded that the totality of available evidence indicated similar efficacy and safety of the biosimilar and the reference product for all therapeutic indications of infliximab.
Epoetin extrapolation
The same working party looked at extrapolation of indications for biosimilar epoetin [9]. Epoetin was an apposite choice, since it has been linked with incidents, albeit rare, of immunogenicity. Recombinant erythropoietin (epoetin) is approved for many indications including the treatment of renal anaemia and chemotherapy-induced anaemia. The rare development of neutralizing, crossreacting anti-epoetin antibodies, which can cause pure red cell aplasia (PRCA), was reported in the late 1990s after the formulation of the originator biological Eprex (epoetin alfa) was changed [10]. Once the cause of this reaction had been established, it was no longer permitted to change the formulation of epoetin without approval by the relevant authorities. Since this ruling, no specific efficacy or safety issues have been identified in clinical practice for biosimilar epoetins licensed in Europe. Post-marketing studies of biosimilar epoetin in the cancer indication have shown it to be well tolerated and efficacious and justify EMA’s decision on extrapolation [9].
Inform physicians
The European Union (EU) was the first highly regulated area to create a legal and regulatory framework for follow-on versions of original innovative biological medicines, similar biological medicinal products (biosimilars) [11]. The biosimilars with valid marketing authorization in the EU are used to treat several types of disease and represent different levels of structural complexity. Sadly, in contrast to the pharmaceutical community, where biosimilars have been intensively debated, physicians have shown little interest in this area, perhaps because they have not been provided with sufficient information.
Professor Pekka Kurki, who chaired EMA’s Committee for Medicinal Products for Human Use (CHMP) Biosimilar Medicinal Products Working Party between 2004 and 2007, has urged regulators in the EU Member States to provide physicians with more information that is already available [11]. Without this reference source, prescribers are left only with information that is influenced by commercial interests, he warns.
Biosimilars worldwide
Problems encountered in the EU on the subject of biosimilars and extrapolation are shared widely, with different countries and regions at different stages of uptake. While science and scientific and therapeutic principles play a role in both interchangeability/substitutability and extra polation of indications, each country has a unique approach, based on national and local laws and practice issues and perceptions. In addition to regulations that govern interchangeability/substitutability and extrapolation of biosimilars, each country or region has different administrative processes regarding the substitution of a prescribed product with another ‘equivalent product’.
Canada has taken a cautious approach to extrapolation of indications and uses. In Canada, where biosimilars are known as subsequent entry biologicals (SEBs), the indication of SEBs must be the same as those of the reference product [12]. This is different to EMA, which covers the extrapolation of uses to all those approved for a class of products.
As knowledge and experience surrounding biosimilars increases, regulatory agencies may be able to overcome the scientific and regulatory hurdles and harmonize their decision-making at a global level [13].
Since FDA did not approve its first biosimilar – Zarxio (filgrastimsndz) – until 2015 [14], extrapolation or interchangeability is a relatively new concept in the US. Under US legislation, interchangeable or interchangeability means that a biological product is biosimilar to the reference product and there is an expectation that the two products will produce the same clinical result in any given patient [14]. According to this ruling, when a given biological product is administered more than once to an individual, the risk of alternating or switching between the biosimilar and its reference product (in terms of safety or efficacy) is no greater than the risk of using the reference product without such alternation or switch. This is clearly a ruling more suited to a generic than a biosimilar medicine, and some US states are moving, or proposing to move, to a vote on amendments to their generic-equivalent laws to include guidance on biosimilars [12].
Summary
With a growing number of biosimilars appearing on the market, and the first approval of a biosimilar in the US [14], clear guidelines on establishing biosimilarity, and approving subsequent extrapolation, are urgently required.
Here, we show that analytics are essential in establishing biosimilarity, since they are typically more sensitive than traditional clinical endpoints. Clinical studies are important, however, because they can be used to confirm biosimilarity once it has been established. This is crucial with regards to testing for immunogenicity, which, although extremely rare, could be life threatening.
The body of knowledge about the clinical relevance of quality attributes is extensive and continues to grow. It remains important to remember that extrapolation is from the reference product to the biosimilar based on the entire similarity exercise, and not from one indication to others.
Editor’s note
Figures and tables are reprinted with permission from Dr Jeorg Windisch.
Competing interests: None.
Provenance and peer review: Article abstracted based on extensive research; externally peer reviewed.
Bea Perks, PhD, GaBi Journal Editor
References
1. GaBI Online – Generics and Biosimilars Initiative. Extrapolation for biosimilars [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Aug 15]. Available from: www.gabionline.net/Biosimilars/Research/Extrapolation-for-biosimilars
2. Mire-Sluis T. The regulatory implications of the ever increasing power of mass spectrometry and its role in the analysis of biotechnology products – where do we draw the line? Available from: http://c.ymcdn.com/sites/www.casss.org/resource/resmgr/Mass_Spec_Speaker_Slides/2012_MS_Mire-SluisTony.pdf
3. Windisch J. Value of Analytical and clinical data in supporting biosimilarity and extrapolation. DIA Biosimilars; 18–19 September 2014; Washington, DC, USA.
4. U.S. Food and Drug Administration. Guidance for industry. Scientific considerations in demonstrating biosimilarity to a reference product. April 2015 [homepage on the Internet]. 2015 Apr 24 [cited 2015 Aug 15]. Available from: http://www.fda.gov/downloads/DrugsGuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
5. European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005 Rev. 1. 18 December 2014 [homepage on the Internet]. 2014 Dec 23 [cited 2015 Aug 15]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf
6. Ramachandra S, Bukofzer S. Why extrapolation is paramount to achieving the full promise of biosimilars. Hospira Policy Paper. April 2015.
7. GaBI Online – Generics and Biosimilars Initiative. Extrapolation of indications in biosimilars: infliximab [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Aug 15]. Available from: www.gabionline.net/Biosimilars/Research/Extrapolation-of-indications-in-biosimilars-infliximab
8. Weise M, et al. Biosimilars: the science of extrapolation. Blood. 2014;124(22): 3191-6. doi:10.1182/blood-2014-06-583617. Epub 2014 Oct 8
9. GaBI Online – Generics and Biosimilars Initiative. Extrapolation of indications in biosimilars: epoetin [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Aug 15]. Available from: www.gabionline.net/Biosimilars/Research/Extrapolation-ofindications-in-biosimilars-epoetin
10. GaBI Online – Generics and Biosimilars Initiative. Epoetin alfa and pure red cell aplasia [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Aug 15]. Available from: www.gabionline.net/Biosimilars/Research/Epoetin-alfa-and-pure-red-cell-aplasia
11. Kurki P. Biosimilars for prescribers. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(1):33-5. doi:10.5639/gabij.2015.0401.008
12. Klein AV, Wang J, Bedford P. Subsequent entry biologics (biosimilars) in Canada: approaches to interchangeability and the extrapolation of indications and uses. Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(3):150-4. doi:10.5639/gabij.2014.0303.033
13. Pen A, Klein AV, Wang J. Health Canada’s perspective on the clinical development of biosimilars and related scientific and regulatory challenges. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(1):36-41. doi:10.5639/gabij.2015.0401.009
14. GaBI Online – Generics and Biosimilars Initiative. FDA approves its first biosimilar [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2015 Aug 15]. Available from: www.gabionline.net/Biosimilars/News/FDAapproves-its-first-biosimilar
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2015 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



Can I have the reference list by return of mail?
Dear Dr Laszlo Varga,
We very much appreciate your kind feedback, the references of the article ‘Supporting biosimilarity and extrapolation’ are uploaded on the website, please continue with your valuable comment to GaBI Journal.
Thank you for your interest in GaBI. Please enjoy the quality information and content published under GaBI (GaBI Online and GaBI Journal)
GaBI Journal Editorial Office