Barriers to market uptake of biosimilars in the US
Published on 2014/07/03
Generics and Biosimilars Initiative Journal (GaBI Journal). 2014;3(3):108-15.
Author byline as per print journal: Joshua P Cohen, PhD; Abigail E Felix, BA; Kim Riggs, MPH; Anumeha Gupta, MD
|
Background: In the US, a new approval pathway for biosimilars has been established as part of the Affordable Care Act. Biosimilars are anticipated to increase treatment options and lower the growth in spending on biologicals. How the commercial prospects for biosimilars will play out in the US is uncertain. From a regulatory, approvals, and market standpoint, Europe is ahead of the US with respect to biosimilars. Lessons may be drawn from European experience. |
Submitted: 17 June 2014; Revised: 8 August 2014; Accepted: 15 August 2014; Published online first: 29 August 2014
Introduction
In 2013, biologicals – medicinal products made by or derived from living organisms – comprised an annual global market of US$170 billion with recombinant insulin, human growth hormone, erythropoietins, and monoclonal antibodies among the leading categories of products. IMS Health estimates that biologicals comprising US$64 billion worth of products will be off patent in the US and European markets by 2015 [1].
As patents for small-molecule pharmaceuticals expire, generic versions enter the market at discounted prices. Development of generic small-molecule drugs has helped to reduce these costs. However, the development of biosimilars has lagged behind small-molecule generics. As biologicals begin to go off patent, the opportunities for expansion of therapeutic alternatives and cost savings will be the primary drivers of biosimilar development and introduction into global markets [2]. In contrast to small-molecule drugs, biologicals are complex, making it impossible to manufacture identical copies; hence, the use of the term ‘biosimilars’ for ‘generic’ versions of biologicals [1, 3]. Here, we define biosimilars as biologicals approved through an abbreviated approval process that references an originator biological in the regulatory submission [4].
The Affordable Care Act establishes an abbreviated licensure pathway for biosimilars. The Biologics Price Competition and Innovation Act (BPCIA) of 2009 was folded into the Affordable Care Act. The BPCIA has analogous objectives for biosimilars as the Hatch-Waxman Act with respect to small-molecule generics. Specifically, the BPCIA aligns the pathway for biosimilar approval with US Food and Drug Administration (FDA) regulations, which allow developers to include information that is already known about the originator product [5]. The BPCIA establishes specific requirements for the development of biosimilars, see Table 1.

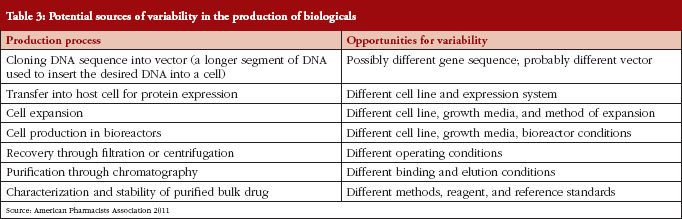
The development of biosimilars is much more challenging than the development of small-molecule generics, due to the greater complexity of biological drugs (chemical structure, analytical characterization) and the complex manufacturing process [6], see Table 2. And it is because of this inherent complexity that the production, approval, and uptake of biosimilars follow a different trajectory than the existing generic drug market, see Table 3.

To date, FDA has received 56 meeting requests for 13 biosimilar products, and 17 Investigational New Drug applications. However, it is noteworthy that no biologics licensing applications (BLAs) have been submitted under the 351(k) biosimilar pathway [7].
The US accounts for most of the global spending on biologwicals, and will therefore be a key driver of long-term biosimilars’ market potential [8]. At present, however, Europe leads the way from a regulatory, approvals, and market uptake standpoint. A European Medicines Agency (EMA) pathway for biosimilar approvals has been in place since 2006. The European experience may offer lessons for how the future will unfold in the US. To date, there are 17 approved biosimilars in the EU, corresponding to five reference products, see Table 4.

Healthcare budget cuts and the accumulation of evidence on the relative safety of approved biosimilars are contributing to more prescribing of biosimilars [1, 3]. In 2010, biosimilars’ overall market share in Europe was 15% and is projected to be over 20% by the end of 2013 [9]. Further, the introduction of biosimilars has resulted in substantial cost savings in Europe. Estimates predict that between 2007 and 2020 the use of biosimilars will result in an overall savings of between Euros 11.8 and Euros 33.4 billion, with cost reductions concentrated in France, Germany, and the UK [10, 11].
How the commercial prospects for biosimilars will play out in the US is uncertain. Market uptake will initially depend on regulatory policies, including the smoothing out of issues concerning the FDA’s regulatory pathway. The key to expediting a shift to biosimilars is establishing interchangeability. In the case of generic drugs, once a product receives an AB rating from FDA – implying that it is bioequivalent to its reference product – a pharmacist is allowed to automatically substitute the generic for the brand-name drug without physician approval, subject to state laws. In order for a biological to be declared interchangeable with its originator or reference product, the manufacturer must show that the product is not only biosimilar, but that it can be expected to produce the same clinical result as the reference product in any given patient [6]. At the payer level, formulary management will have a substantial impact on biosimilar adoption. Once marketing authorization is granted to biosimilars, payer pressure will likely drive market uptake because of the lower costs of biosimilars. At the biosimilar manufacturer level, commercialization support, including physician education and patient co-payment assistance, will drive adoption. Additionally, originator manufacturers will continue to lobby at the state level to make automatic substitution by pharmacists of branded biologicals more cumbersome. Finally, market uptake will depend on the relative influence of prescribing physicians, and the degree to which patients express a preference for biosimilar over originators.
Between 2006 and 2012 biosimilar sales have nearly doubled from US$6.4 billion to US$12.4 billion [12]. Nevertheless, the market uptake of biosimilars is not occurring at the predicted rates. For example, within two years of launch, biosimilar versions of erythropoetins gained 37% of the market share in Europe. Compare this with a typical generic drug, which in the US accounts for 90% of market share within one year of entry [5].
There are multiple reasons for the slower than expected uptake in Europe, including the lack of automatic substitution of biosimilars for originator products, and the fact that many doctors and patients are reluctant to switch or substitute, given their lack of familiarity with biosimilars [13]. Furthermore, large differences exist and persist in biosimilar penetration between various European markets, with Germany, the UK, and Italy leading the way. Germany’s comparatively high uptake may be explained in part by establishment of a quota system, in which specialists must prescribe new patients a certain percentage of biosimilars relative to biologicals. Also, reference pricing, or reimbursement limits calculated per therapeutic class, above which consumers must pay a surcharge, has played a role in supporting growth of biosimilar sales. Despite suboptimal market uptake thus far, there is evidence of positive financial impact of biosimilar use in Europe. Projections estimate the use of biosimilars in Europe to result in an overall savings of between Euros 11.8 billion and Euros 33.4 billion between 2007 and 2020, with the largest savings in the UK, France and Germany [10].
In this paper, we examine challenges and opportunities with respect to market uptake of biosimilars in the US, from the perspectives of payers and physicians. Section II describes the methods we used to conduct our study. Section III reports the study’s findings. Section IV discusses policy implications related to the study’s main findings.
Methods
In order to examine challenges and opportunities with respect to market uptake of biosimilars, we first conducted a review of Medline-indexed publications using the terms ‘biosimilar’ and ‘biologic’. The search was limited to the past five years (2008 to present), and only included items with abstracts, written in English. Twenty-one publications were selected (from a total of 50 that were sourced) for full-text review based on the relevance to the research question and their usefulness in informing the development of the physician and payer surveys. In addition, similar searches were conducted using Internet search engines to identify non-Medline-indexed articles, such as grey literature sources and white papers.
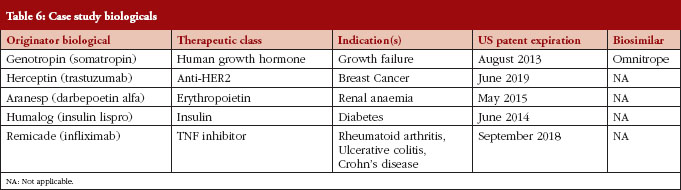
Following our literature review, we developed two stakeholder surveys. For each stakeholder, a distinct web-based survey instrument was developed, targeting specific topics of specific relevance for the stakeholder in question, as outlined in Table 5. We used case studies to elicit attitudes towards specific biologicals likely to be the first biosimilars available, see Table 6. The specific compounds for the case studies were selected by identifying biologicals that: (1) had patent expiration dates in the next two to 10 years; (2) comprised a large portion of global pharmaceutical sales; and (3) represented a diversity of indications.

Payers were selected as a key stakeholder because they are anticipated to be a driving force behind biosimilar adoption. Biosimilars will likely offer price discounts of between 15% and 35% compared with the originator products [14]. In turn, competition will drive down prices of originator products.
We selected 36 payers from the Tufts CSDD (Center for the Study of Drug Development) database of contacts from previous surveys. Furthermore, we conducted a Medline/Scopus search to identify payers who had written on or were familiar with biosimilars. Note, relative to the universe of payers there are comparatively few who are familiar with biosimilars. Eight responded (24% response rate). Payer respondents represent eight of the top 25 in terms of numbers of covered lives.
Physicians were selected as the second key stakeholder group due to their direct involvement in the prescribing of pharmaceuticals. Generally, physicians are relatively conservative prescribers, and slow to adopt new technologies. Also, most physicians in the US are unfamiliar with biosimilars. We therefore conducted a Medline/Scopus search to identify 42 physicians who had written on or were familiar with biosimilars. Fourteen responded with completed questionnaires (33% response rate). The respondents were specialized in nephrology, oncology, dermatology, or rheumatology.
The surveys were designed to be qualitative in nature, specifically looking to capture attitudinal data through the use of Likert scales. From our literature review, we determined that many physicians and payers are unaware of biosimilars. Our goal was to identify barriers to prescribing and uptake that exist among those who are aware of biosimilars. Hence, our ‘selection bias’ is intentional.
Findings
Literature review
We identified merely a handful of US-based research articles collecting views of payers and physicians on biosimilar uptake. The most robust was a study conducted at the 2011 National Comprehensive Cancer Network Annual meeting. The 4-question survey was made available to attendees at the conference. In this survey, researchers focused on four broad categories: (1) familiarity with biosimilar legislation; (2) interest in prescribing, dispensing and administering biosimilars; (3) importance of various types of information; and (4) anticipated use of biosimilar products for specific classes of biologicals [15].
Two hundred and seventy-seven conference attendees responded to the survey (response rate of less than 5%). Most respondents were physicians (n = 129), followed by nurses (n = 71), pharmacists (n = 38), and other types of clinicians (n = 39). Overall, 36% of respondents indicated they were not at all familiar with biosimilars and the recent legislation to establish an approval pathway. Despite a lack of familiarity with biosimilars, a majority of respondents expressed high (27%) or moderate (35%) interest in prescribing, dispensing or administering biosimilars in their practice settings. Of the types of information listed in the survey, a majority of respondents listed them all as ‘very important’ to their decision-making process, the one exception being ‘colleague and expert opinion’, see Figure 1.

Responses to the question, ‘As more information on biosimilars becomes available, how important are the following types of information in helping you decide to use biosimilar products? [15].
Irrespective of the type of biological reference product, a majority of respondents indicated they ‘would require review and discussion’ before using an FDA-approved biosimilar. This finding contradicted the researchers’ hypothesis that biosimilar agents for supportive care indications would be more readily used than those indicated for the active treatment of cancer. This suggests a deeper level of inquiry is needed to fully understand physician decision making with regard to biosimilars [15].
Dranitsaris et al. highlight cost savings as the main driver for uptake of biosimilars from the payer perspective [4]. However, the paper states that physicians are unlikely to prioritize cost savings over patient preferences and outcomes. Some of the specific challenges Dranitsaris et al. identified that manufacturers may face following FDA approval include the implementation of pharmacovigilance programmes, patient and physician acceptance, commercial scale-up, intensity of competition, and level of price erosion. This list of challenges acknowledged by Dranitsaris suggests the complexity of both the emerging biosimilar market and the multiple stakeholder groups that need to be considered – not only payers and physicians, but also manufacturers and patients.
A research firm examined payer attitudes toward biosimilars, specifically looking at cost discounting expectations [16]. Findings suggest that payers in a managed care network expect a 10% to 20% discount from the branded price. But, payers stated that if they were going to institute a mandatory policy of switching existing patients from the branded product to a biosimilar, they would require a 40% cost savings. In addition to providing valuable insights into cost expectations of payers, this survey pointed to trends in payer attitudes. For example, in September 2008, 50% of payers said that an ‘official equivalency designation’ rating for biosimilars was ‘absolutely necessary’ in order to incorporate biosimilars into coverage plans. However, in November 2011, only 33% said this designation was absolutely necessary.
Several sources identified in the literature review provided revealing quotes from payers on their attitudes towards biosimilar market uptake, see Table 7 [17–19].

Results from a recent pan-European survey [20, 21] suggest physicians have limited knowledge of biosimilars. Fifty-four per cent claimed to have a basic understanding. However, 24% could not define or had not heard of biosimilars before. Additionally, only 22% considered themselves very familiar. Likewise, another recent survey [22] also indicated a low level of awareness among specialist physicians. To illustrate, only 8% of rheumatologists surveyed knew that there were biosimilars in the development pipeline for rheumatoid arthritis.
The published literature summarizing European surveys of physicians identifies challenges and opportunities for biosimilar market uptake in the US [23]. Challenges include lack of familiarity and uncertainty by key stakeholders, opposition by brand (originator) biological developers, and preemptive legislation being passed by states. The main challenge for biosimilars extends beyond obtaining approval by FDA through the abbreviated approval pathway. FDA approval is a necessary but insufficient condition. Biosimilars will also need to gain market access and market share relative to originator products. At the same time, biosimilars present opportunities to payers and physicians, including cost containment and increased availability of therapeutic options. In order to assess the attitudes of payers and physicians in the US we designed surveys. The surveys will help identify challenges and opportunities as perceived by payers and physicians.
Survey results
Physicians
Almost all physicians surveyed believe that if a biosimilar is approved by FDA the product will perform similarly to the originator biological with regard to safety and efficacy. Most (70%) physicians say they are likely to prescribe biosimilars to a new patient, given the current state of regulations and knowledge with respect to biosimilars, see Figure 2. The majority of physicians also feel comfortable switching an existing patient from the originator biological to a biosimilar, see Figure 3.


Physicians were asked: Assuming similar efficacy and safety, how likely would you be to prescribe a biosimilar to a new patient who has not been previously treated for their condition?
Physicians were asked: How likely they would be to switch an existing patient from the originator to a biosimilar?
FDA may soon classify a biosimilar as ‘interchangeable’ with the originator. In addition to demonstrating biosimilarity, a manufacturer must show that the proposed interchangeable product is expected to produce the same clinical results in terms of safety and efficacy as the originator, see Figure 4.

Physicians were asked: If an interchangeable biosimilar were available for the originator you normally would prescribe, how likely would you be to prescribe this product to new patients?
From our survey it appears that efficacy and safety are the two most important considerations that influence a physician’s decision to prescribe a biosimilar. Out-of-pocket costs to patients, price of treatment, and immunogenicity have less influence on a physician’s decision. Fifty per cent of physician respondents consider it ‘very important’ that there be proven chemical and pharmacokinetic similarities between originators and biosimilars. Roughly, half of respondents considered payer and cost considerations ‘very important’.
Almost all physicians are in favour of implementing coverage with evidence development programmes as a way to assess post-marketing safety and effectiveness while ensuring (new) patient access to biosimilars: prescribing and coverage of a biosimilar following FDA approval, provided patients enroll in post-approval clinical trials to assess real-world effectiveness and safety.
Payers
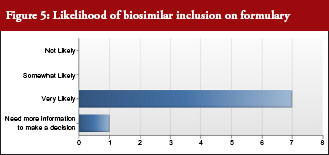
All but one payer intended to include biosimilars on their formulary, see Figure 5. In addition, all payers intended to promote use of biosimilars by differentiating between the originator biological and biosimilar through the use of co-payment or co-insurance tiering to steer patients and physicians towards biosimilars. Notably, half of payer respondents would include European evidence as evidence supporting formulary decisions.
All payers would recommend biosimilars to new patients. Several would require that new patients take biosimilars first. All would promote biosimilar prescribing, and all would steer patients towards biosimilars through the use of formulary management tools. All payers anticipated switching of patients from originator to biosimilar within one year of launch of biosimilar. Payers are more comfortable with interchangeability designation for older products, such as erythropoetins.
It is notable that half of payer respondents were reluctant to institute the practice of automatic substitution of biosimilars for originators. Most payers would recommend use of a biosimilar to treat a condition for which it is not specifically approved, but for which the originator has a labelled indication.
Payers were asked: Once approved by FDA, what is the likelihood that you would include biosimilars on your formulary?
Safety and efficacy were the most important factors when considering adoption of biosimilars on formularies. Cost-effectiveness and out-of-pocket costs to patients were the least important considerations.
Seventy-five per cent of payer respondents said they would recommend therapeutic switching of biosimilars, see Figure 6. All payer respondents declared there would be automatic therapeutic switching of biosimilars across all therapeutic classes at some point in the near future. And, a majority of payers supported extrapolating the use of a biosimilar to an indication for which it is not approved, but for which the originator biological has a labelled indication. The older the class of biologicals, the more readily payers appeared to support extrapolating use of a biosimilar for an indication for which it is not approved.
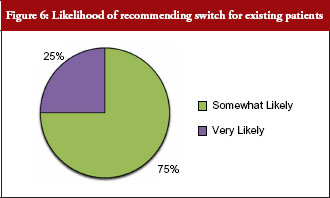
Payers were asked: How likely would you be to recommend the biosimilar to existing patients, i.e. switching?
Next, we asked payers whether they would recommend use of a biosimilar to treat a condition for which it is not specifically approved, but for which the originator has a labelled indication. This implies extrapolating the use of a biosimilar to an indication for which clinical trial data have not been submitted for FDA approval, but for which the originator is approved, see Figure 7. The majority recommended biosimilars for unlabelled uses.

Almost all payers anticipated that growth hormone products would be eligible for therapeutic switching first, followed by insulins, and then erythropoiesis stimulating proteins, while tumour necrosis factor-α blockers and human epidermal growth factor receptor (HER2) inhibitors would be switched last. This is consistent with the level of comfort with the interchangeability designation, with most feeling comfortable with an interchangeability designation for insulins and growth hormone products, but only two feeling comfortable with tumour necrosis factor-α blockers and human epidermal growth factor receptor (HER2) inhibitors.
Seventy-five per cent of payer respondents expect biosimilars to have a 15–35% price discount. The remainder of respondents thinks a larger discount is likely. The highest discount is expected for growth hormone products, and the lowest for HER2 inhibitor products.
Discussion
In sum, for both payers and physicians, the most important considerations were safety and efficacy, followed by out-of-pocket cost to patients, immunogenicity, and price of treatment. Both physician and payer respondents would distinguish between treatment-naïve patients and those who are already on an originator biological. They cited risk for patients already benefitting from originator therapy. As a result, physicians and payers feel more comfortable prescribing a biosimilar for treatment-naïve patients rather than switching from an originator product.
Market uptake will depend on regulatory policies, including the smoothing out of issues concerning FDA’s regulatory pathway [24]. At the payer level, formulary management will have a major impact on biosimilar adoption. At the biosimilar manufacturer level, commercialization support – including physician education and patient co-payment assistance – will drive adoption. Finally, market update will depend on the relative infl uence of prescribing physicians, and the degree to which patients express a preference for biosimilar over originator products [25].
Physicians and payers will play a key role with respect to uptake of biosimilars. Physicians and payers will likely display caution when deciding on prescribing biosimilars to existing patients, i.e. switching. They will look to regulatory authority guidance for further support. Specifically, there needs to be more regulatory clarity on interchangeability. Recently published FDA guidance on biosimilars is expected to clarify the kind of clinical pharmacology data necessary to demonstrate interchangeability.
Interchangeability will be a key driver of biosimilar utilization, and serve as the basis for state pharmacy substitution laws. To maximize cost savings, payers will likely employ formulary management tools, such as higher cost-share tiering for originator products and lower cost sharing for biosimilars.
The expected price discount for biosimilars is not large. Furthermore, higher rebates on originator biologicals may be used by manufacturers as a barrier to adopting biosimilars. Such rebates will likely minimize the cost differential between biosimilars and originators. Therefore, biosimilar manufacturers will likely have to treat them as any other branded product. Furthermore, initially biosimilars will likely compete as non-interchangeable therapeutic alternatives. Finally, biosimilars will be subject to dynamic competition from new biologicals in the same therapeutic class – including incremental improvements to existing originator products [26].
To ensure access to and monitoring of post-marketing safety and effectiveness of biosimilars, payers may wish to establish patient registries through coverage with evidence development arrangements.
Disclosure of financial and competing interests: Tufts Center for the Study of Drug Development received a grant from Sandoz to conduct this study. The authors acknowledge their gracious support.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Joshua P Cohen, PhD
Abigail E Felix, BA
Kim Riggs, MPH
Anumeha Gupta, MD
Tufts Center for the Study of Drug Development, Tufts University, Suite 1100, 75 Kneeland Street, Boston, MA 02111, USA
References
1. Greer F. Biosimilar development: the race to market continues. SGS Life Science. 2013;45.
2. Comes P. The economic pressures for biosimilar drug use in cancer medicine. Target Oncol. 2012;7 Suppl 1:S57-67.
3. Tsiftsoglou, et al. Development and regulation of biosimilars: current status and future challenges. BioDrugs. 2013;27(3):203-11.
4. Dranitsaris G, Amir E, Dorward K. Biosimilars of biological drug therapies: regulatory, clinical, and commercial considerations. Drugs. 2011;71(12):1527-36.
5. American Pharmacists Association. The biosimilar pathway: where will it lead us? Pharmacy Today. 2011;67-76.
6. Grabowski HG, Guha R, Salgado M. Regulatory and cost barriers are likely to limit biosimilar development and expected savings in the near future. Health Aff (Millwood). 2014;33(6):1048-57.
7. McCaughan M, Gingery D. Where are the biosimilars? The RPM Report. 2013;30-1.
8. IMS Market Prognosis: May 2012.
9. IMS MIDAS/MTA Global Database: March 2011.
10. Haustein R, de Millas C, Hoer A, Haussler B. Saving money in the European healthcare systems with biosimilars. Generics and Biosimilars Initiative (GaBI Journal). 2012;1(3-4):120-6. doi:10.5639/gabij.2012.0103-4.036
11. Vogler S, Zimmermann N. The potential of generics policies: more room for exploitation–PPRI Conference Report. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(3-4):146-9. doi:10.5639/gabij.2012.0103-4.030
12. GaBI Online – Generics and Biosimilars Initiative. Biologics sales have almost doubled since 2006. [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2014 Aug 8]. Available from: www.gabionline.net/Biosimilars/General/Biologicals-sales-have-almost-doubled-since-2006
13. GaBI Online – Generics and Biosimilars Initiative. Biosimilars approved in Europe [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2014 Aug 8]. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe
14. Blackstone E, Fuhr J. The economics of biosimilars. American Health & Drug Benefits. 2013;6(8):1-14.
15. Zelenetz AD, Ahmed I, Braud E, et al. NCCN Biosimilars White Paper: regulatory, scientific, and patient safety perspectives. J Natl Compr Canc Netw. 2011;9 Suppl 4:S1-22.
16. Xcenda. Payer perspectives on biosimilars [homepage on the Internet]. 2014 Aug 8 [cited 2014 Aug 8]. Available from: http://www.xcenda.com/Insights-Library/Payer-Perspectives/Payer-Perspectives-on-Biosimilars/
17. Thompson CA. Forum discusses biosimilars, better biologics. Am J Health Syst Pharm. 2011;68(23):2210.
18. Senior M. Biosimilars battle rages on, Amgen fights both sides. Nat Biotechnol. 2013;31(4):269-70.
19. Reink T. Biosimilars might not measure up to health plan expectations. Manag Care. 2012;21(10):12-3.
20. Alliance for Safe Biologic Medicines. EuropaBio joins the Alliance for Safe Biologic Medicines. 27 June 2013 [homepage on the Internet]. 2014 [cited 2014 Aug 8]. Available from: http://safebiologics.org/resources/2013/06/europabio-joins-the-alliance-for-safe-biologic-medicines-asbm/
21. Dolinar RO, Reilly MS. Biosimilars naming, label transparency and authority of choice – survey findings among European physicians. Generics and Biosimilars Initiative Journal (GaBI Journal). 20143;3(2):58-5. doi:10.5639/gabij.2014.0302.018
22. PMLiVE. Biosimilars: friend or foe for healthcare systems? Available from: http://www.pmlive.com/pharma_intelligence/biosimilars_friend_or_foe_for_healthcare_systems_547599
23. BBC Research. Biologic therapeutic drugs: technologies and global markets. January 2011. Report code BIO079 A [homepage on the Internet]. [cited 2014 Aug 8]. Available from: http://www.bccresearch.com/market-research/biotechnology/biologic-therapeutic-drugs-bio079a.html
24. Greer F. Biosimilar development: from science to market. Life Science. 2011;47.
25. Marwood Business Group. Washington Healthcare Report. March 2014 [homepage on the Internet]. 2014 Mar 20 [cited 2014 Aug 8]. Available from: http://www.marwoodgroup.com/wp-content/uploads/2013/11/The-Washington-Healthcare-Report_ABSTRACT_March-2014.pdf
26. GaBI Online – Generics and Biosimilars Initiative. Germany’s rational use of medicines [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2014 Aug 8]. Available from: www.gabionline.net/Reports/Germany-s-rational-use-of-medicines
|
Author for correspondence: Research Associate Professor Joshua P Cohen, PhD, Tufts Center for the Study of Drug Development, Tufts University, Suite 1100, 75 Kneeland Street, Boston, MA 02111, USA |
Copyright © 2014 Pro Pharma Communications InternationalDisclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.



Regarding the Felix et al article – Barriers to market uptake of biosimilars in the US. I’m interested in citing this well researched article. Is it possible to obtain a PDF of the whole article with tables and references? Thank you.
Dear Dr Anne Gentry,
We very much appreciate your kind feedback. Article PDF is available on the website for free download.
Thank you for your interest in GaBI. Please enjoy the quality information and content published under GaBI (GaBI Online and GaBI Journal).
GaBI Journal Editorial Office