First ASEAN educational workshop on regulation and approval of biosimilars/similar biotherapeutic products 2017 – Report
Published on 2018/06/14
Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(3):127-32.
Author byline as per print journal: Robin Thorpe, PhD, FRCPath; Elwyn Griffiths, PhD, DSc; Niklas Ekman, PhD
|
Abstract: |
Submitted: 19 April 2018; Revised: 12 July 2018; Accepted: 18 July 2018; Published online first: 25 July 2018
Introduction
Similar Biotherapeutic Products (SBPs or biosimilars) are being increasingly developed worldwide. The presence of this new class of biotherapeutic agent is improving access and availability to treatment for patients across the globe. However, the regulation and approval of biosimilars is not straightforward and practices differ from country to country. To ensure that biosimilars successfully enter markets and maintain the safety and efficacy achieved by their originator products, methods for their approval and regulation need to be laid out clearly.
To discuss the regulation and approval of biosimilars across the nations of Southeast Asia, the First ASEAN Educational Workshop on Regulation and Approval of Biosimilars/Similar Biotherapeutic Products took place on 23 July 2017, Bangkok, Thailand [1]. This was organized by the Generics and Biosimilars Initiative (GaBI) in collaboration with the Association of Southeast Asian Nations (ASEAN). Forty-three participants, including the speakers, attended the workshop.
This First ASEAN Educational Workshop on Regulation and Approval of Biosimilars/Similar Biotherapeutic Products was an interactive workshop that focused on the regulation and approval of biosimilars. It brought together regulators and key representatives from the ASEAN ACCSQ–PPWG (ASEAN Consultative Committee for Standards and Quality Pharmaceutical Product Working Group) Member States: Indonesia, Lao PDR, Malaysia, Myanmar, Philippines, Singapore, Thailand, Vietnam; along with academics, medical practitioners/specialists, pharmacists, and procurement specialists. These participants held discussions and exchanged information with experts from across Asia, Europe and the US.
One of the key aims of the educational workshop was to address the potential differences in quality as well as the therapeutic and immunological (immunogenicity) effects of biologicals across ASEAN nations. To facilitate this, representatives from regulatory authorities, academia and medical specialists, that are involved in biological and biosimilar development and approval in ASEAN Member States, were joined by expert speakers from other nations. The participants engaged in active discussions covering topics concerning: the principles and challenges related to manufacturing process development of biosimilars; the clinician’s experience of biocopy* erythropoietin (EPO); the regulatory experience of re-evaluating EPO; and the identification of future educational requirements.
Methods
The format of the First ASEAN Biosimilars Educational Workshop was similar to that followed in prior educational workshops as reported in GaBI Journal [2–4]. For more details of methods and case presentations, see the published report of the First Latin American educational workshop on similar biotherapeutic products [2], the First MENA educational workshop on regulation and approval of similar biotherapeutic products/biosimilars [3], and the First Turkish interactive workshop on regulation and approval of similar biotherapeutic products/biosimilars [4].
Expert speaker presentations
The ASEAN workshop began with presentations given by expert speakers [1]. The details of most of these presentations were updates to those given at other GaBI workshops and are summarized in their published reports [2–5].
Dr Thijs J Giezen, a hospital pharmacist at the Foundation Pharmacy for Hospitals in Haarlem, The Netherlands, and a member of the Biosimilar Medicinal Products Working Party (BMWP) of the European Medicines Agency, discussed the safety assessment and risk management of biosimilars, as reported in [2, 5]. However, in this more recent 2017 presentation, he highlighted that in many nations across the globe, patients and physicians have a knowledge gap when it comes to biosimilars. He advised that globally, further steps need to be taken to improve education and to increase trust in biosimilars.
Biocopy* EPO’s in Thailand
In addition to the presentations that have been given at previous GaBI workshops, two ASEAN-specific presentations were given. Professor Kearkiat Praditpornsilpa of Chulalongkorn University, Bangkok, Thailand gave a presentation entitled, ‘Clinician’s experience of biocopy EPO in Thailand’. Here, he discussed the well-documented immunogenicity of recombinant human erythropoietin (r-HuEPO) by subcutaneous exposure. This adverse immunological effect causes anti-r-HuEPO-associated pure red cell aplasia (PRCA). Professor Praditpornsilpa highlighted the fact that there have been increasing cases of anti-r-HuEPO-associated PRCA by subcutaneous exposure in Thailand. It is suggested that the immunogenicity may relate to human leukocyte antigen (HLA) immunogenetics, protein aggregation, stability during storage; and handling of drug products, formulation and drug product quality. Professor Praditpornsilpa and his research group have carried out extensive investigations into the unreliability of biocopy EPO’s in Thailand.
r-HuEPO’s have been licensed for the treatment of renal anaemia in Thailand and include innovator products and more than 20 biocopy products. However, there are inconsistencies in product characterization and quality which may lead to different immunogenicity and safety profiles. As such, the Prospective Immunogenicity Surveillance Registry of r-HuEPO with subcutaneous exposure in Thailand estimated the incidence rate of anti-r-HuEPO associated PRCA among subjects who had subcutaneous exposure to any currently marketed r-HuEPO product. In addition, the registry addressed the risk of anti-r-HuEPO associated PRCA and the association of product quality with adverse immunogenicity.
Following their investigation, the registry suggested that there is an association between product quality and immunogenicity. All anti-r-HuEPO associated PRCA cases had received biocopy r-HuEPO products, resulting in the observation of a correlation between product characteristics and adverse immunogenicity. As patients’ safety is a top priority, Professor Praditpornsilpa emphasized that the approval process for biosimilars in Thailand needs to include intensive scientific regulation. The regulatory pathway for similar biological drugs must be designed to assess quality, characterization and the impurity profile of such products, including head-to-head evaluation with a reference biological product, followed by careful head-to-head evaluation of the non-clinical and clinical aspects. Pharmacovigilance studies should be the final step to ensure quality and safety and this can be assessed together with the cost-effectiveness and patient benefits that such products offer. Professor Praditpornsilpa concluded that, in light of the issues encountered with the use of biocopy r-HuEPO in Thailand, individual case-by-case review and approval of biocopy products is needed.
Re-evaluating erythropoietin’s in Thailand
Mr Pramote Akarapanon, Head of Biological Products Section at the Ministry of Public Health, Thailand, gave a presentation entitled, ‘Experience of re-evaluating erythropoietin in Thailand’. Here, he noted that in Thailand, erythropoiesis stimulating agent EPO’s have been used for the standard treatment of anaemia in patients with chronic kidney disease for some time. However, as mentioned in Professor Praditpornsilpa’s presentation, this was associated with a high prevalence of PRCA. As a result, a re-evaluation of marketed EPO’s was instigated by a Ministerial Order, whereby the importer or manufacturer of EPO is required to submit additional data (quality, non-clinical, clinical) to the Thai Food and Drug Administration (TFDA), together with a Risk Management Plan (RMP). There are two types of RMP depending on the extent of documentation already submitted for each individual product. Mr Akarapanon noted that close monitoring of RMP’s is a big challenge for TFDA.
Summary of the discussions that followed the speakers presentations
Following the speakers presentations, there was the opportunity for discussion of the topics covered. Herein, Dr Niklas Ekman, member of the Biosimilar Medicinal Products Working Party (BMWP) of the European Medicines Agency (EMA), made it clear that EMA only evaluates biosimilars for biosimilarity, and does not evaluate the interchangeability between a biosimilar and reference product. According to the European definition, interchangeability refers to the possibility of exchanging one medicine for another medicine that is expected to have the same clinical effect [6]. He noted that interchangeability is a national concern and is to be dealt with by individual medicines regulatory agencies of the European Union Member States, most of which have published their own position papers or statements on interchangeability that are in general, aligned. He stressed that in Europe, interchangeability means that it is the prescriber that can choose between the originator and the biosimilar, but currently there is no pharmacy-level substitution.
When it comes to switching between an originator and biosimilar, Dr Giezen advised that in Europe EMA, does not require switching studies which is in contrast to the requirements of the US Food and Drug Administration (FDA). In Europe, if a biosimilar is approved it is up to the Member States to decide on interchangeability and switching, not EMA. In general, if a biosimilar and the reference product have been shown to be similar through quality characterization and this confirmed by clinical trials, they can be switched safely and efficaciously. As such, switching studies are not necessary, and are generally only carried out to provide data to show clinicians that switching is safe. Dr Giezen added that switching studies generally have limitations, particularly if the number of patients in the study is small and adverse effects are rare. The design of switching studies and the length of time over which they are carried out, also needs to be considered.
The discussion steered towards understanding how many batches of reference product are needed to establish the Quality Target Product Profile (QTPP) of a biosimilar. Here, Dr Ekman noted that the exact number of batches is not specified by EMA requirements. However, companies are recommended to analyze reference product batches over a relatively long period of time as this means that batch-to-batch variability is better understood. Furthermore, it is in the company’s interest to analyse a reasonable number of batches of the reference product as with a small number of batches the variability present in the quality attribute of the reference product cannot be appropriately estimated. Depending on the quality attribute in question, this can result in analyses of 20 or more reference product batches.
Dr Ekman explained that for evaluating biosimilarity, orthogonal analytical methods should be used whenever possible. If only one method is applied for analysing a certain quality attribute this will usually add more burden on the qualification of the method. It needs to be demonstrated with high confidence that such a method is sensitive and can reliably detect minor differences between the originator and biosimilar. The use of orthogonal methods will increase trust in the biosimilar assessment. Speaker Dr Sundar Ramanan added that, in the case of glycosylation, it is very important to know what kind of enzyme reagents are used in the analysis and that it is critical to assess whether the results obtained from using these enzymes and subsequent separation methods, have the required sensitivity needed for adequate assessment.
In follow up to this, a question was asked about the charge variant analysis that should take place when evaluating a biosimilar. In response, Dr Ekman noted that differences in the charge variant often mean that there are differences between the originator and the biosimilar and it is critical to understand why these differences occur. After this, a more detailed discussion on charge variants and isoforms ensued.
During Professor Praditpornsilpa’s presentation he noted that while investigating r-HuEPOs, his research group encountered a cold chain and storage problem early on in their research. However, when they carried out their pharmacovigilance study, they made sure that the cold chain and storage problem did not interfere with their results. Professor Praditpornsilpa noted that, on inspection of their analytical data, although the human serum albumin was a better stabilizer compared to polysorbate 80, the purity of human serum albumin could be an issue for product quality. This highlights the fact that the quality of the preservation of a biological is key. Professor Praditpornsilpa also noted that he is skeptical about the quality of human serum albumin currently used in the biocopy EPOs in Thailand. He suspects that there may be some contamination but notes that carrying out analysis to confirm this is complicated. The analysis of the products by mass spectroscopy identified other human proteins and also rodent protein contamination. The human proteins could be related to the purity of human serum albumin for preservation and the rodent protein could be related to the separation of r-HuEPO from the cell culture. With regards to the immunogenicity occurring in Thailand that has led to 30 cases of PRCA in 2016, Professor Praditpornsilpa confirmed that this mostly occurred when biocopy products were used, rather than with originator products (although 1−2 cases per year are seen with originator products). He and his team believe that this issue is caused by protein aggregation and impurity of products.
Mr Pramote Akarapanon’s presentation also focused on PRCA occurring following EPO use. Due to the safety concerns surrounding this form of medication in Thailand, he noted that products associated with causing PRCA should be withdrawn from the market. However, during the discussion, Mr Akarapanon noted that there are no specific set timelines in place for product withdrawal and that products were being assessed on a case-by-case basis. In contrast, WHO Guideline on the reassessment of approved rDNA-derived biotherapeutics proposes a stepwise approach which includes timelines for undertaking the exercise. He also added that the public were aware of the safety concerns and of the fact that EPO products are under review.
Parallel case study working sessions
Case study working sessions took place following the presentations (downloadable from the GaBI website [1]). These followed the same format as those described in [2–4], of two fictional cases of a follow-on biological therapeutic, one an erythropoietin product and the other a monoclonal antibody. The participants were divided into four discussion groups, see Table 1, where they evaluated the fictional data supplied. The group discussions are summarized in Tables 2 and 3.

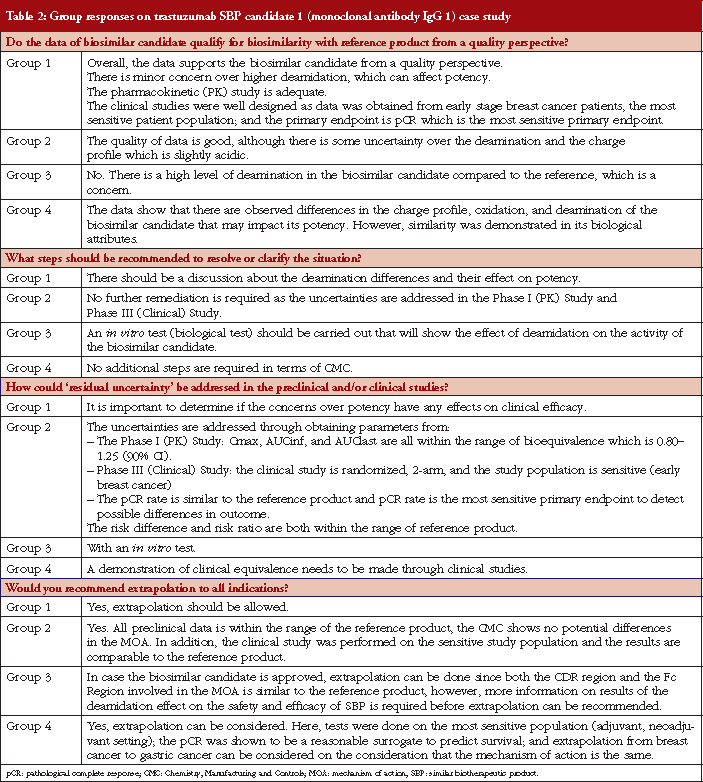
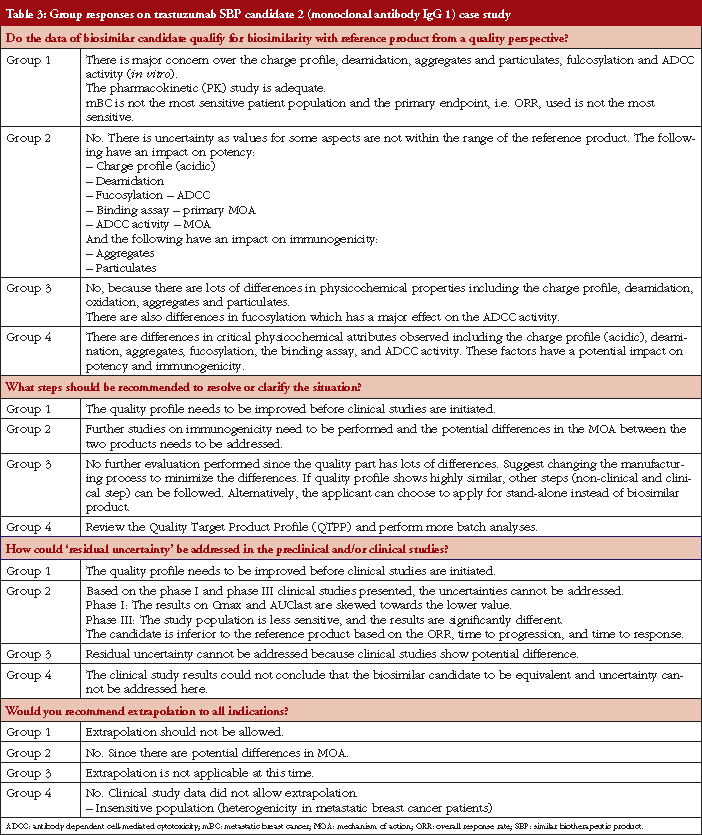
Conclusion
Biosimilar medicines are being increasingly developed and used across Southeast Asia. Safety and efficacy issues that have surrounded the use of biocopy EPO’s in Thailand highlight the need for adequate evaluation and regulation of biosimilars across the region. The ASEAN Educational Workshop on Regulation and Approval of Biosimilars/Similar Biotherapeutic Products was successful in bringing representatives from ASEAN nations together with those from Europe and the US, to discuss the best routes forward for successful biosimilar approval, regulation and market introduction.
Speaker Faculty and Moderators
Speakers
Pramote Akarapanon, BSc Pharm, MA, Thailand
Niklas Ekman, PhD, Finland
Thijs J Giezen, PharmD, MSc, PhD, The Netherlands
Elwyn Griffiths, PhD, DSc, UK
Professor Andrea Laslop, MD, Austria
Professor Kearkiat Praditpornsilpa, MD, Thailand
Sundar Ramanan, PhD, USA
Robin Thorpe, PhD, FRCPath, UK
Meenu Wadhwa, PhD, UK
Moderators
Agnes Chan, Singapore
Azizah Ab Ghani, PhD, Malaysia
Associate Professor Maneewan Suksomtip, PhD, Thailand
Assistant Professor Wisit Tangkeangsirisin, PhD, Thailand
Editor’s comment
All moderators had provided the discussion/conclusion of the group discussion, read the report and revised the content of the summary discussion.
Acknowledgement
The Generics and Biosimilars Initiative (GaBI) wishes to thank Ms Sylvia Laksmi Sardy and Ms B Lusia Herwahyu S from the ASEAN Secretariat for their support towards the coordination of the workshop with ASEAN member states; Dr Tankamol Chanprapap, Ms Charunee Krisanaphan and Mr Pramote Akarapanon of Thai FDA for their strong support through the offering of advice and information towards the preparation and organization of the workshop; the moderators in clarifying the information of the case study discussion when finalizing the meeting report; as well as Dr Robin Thorpe and Professor Andrea Laslop, chair and co-chair of the 2017 workshop; for their strong support through the offering of advice and information during the preparation of the workshop.
The authors would like to acknowledge the help of all the workshop speaker faculty and participants, each of whom contributed to the success of the workshop and the content of this report, as well as the support of the moderators and co-moderators: Ms Agnes Chan, Dr Azizah Ab Ghani, Associate Professor Maneewan Suksomtip and Assistant Professor Wisit Tangkeangsirisin in facilitating meaningful discussion during the parallel case study working sessions, presenting the discussion findings at the workshop, and contributing in the finalization of this meeting report.
Lastly, the authors wish to thank Ms Alice Rolandini Jensen, GaBI Journal Editor, in preparing and finalizing this meeting report manuscript and providing English editing support on the group summaries.
Competing interests: The workshop was sponsored by an unrestricted educational grant to GaBI from Amgen Inc.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Robin Thorpe, PhD, FRCPath, UK
Elwyn Griffiths, PhD, DSc, UK
Niklas Ekman, PhD, Finland
*Biocopy refers to a licensed product made as a copy of an original product but not licensed according to the now accepted international norms of a biosimilar.
References
1. Generics and Biosimilars Initiative. First ASEAN Educational Workshop on Regulation and Approval of Biosimilars/Similar Biotherapeutic Products 2017. 23 July 2017; Bangkok, Thailand. Available from: www.gabi-journal.net/about-gabi/educational-workshops/first-asean-educational-workshop-on-regulation-and-approval-of-biosimilarssimilar-biotherapeutic-products-2017
2. Walson P, Thorpe R. First Latin American educational workshop on similar biotherapeutic products, Mexico City, Mexico, 20 January 2015. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(3):143-8. doi:10.5639/gabij.2015.0403.031
3. Walson P, Thorpe R. First MENA educational workshop on regulation and approval of similar biotherapeutic products/biosimilars, Dubai, United Arab Emirates, 1 September 2015. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(4):173-7. doi:10.5639/gabij.2015.0404.039
4. First Turkish interactive workshop on regulation and approval of similar biotherapeutic products/biosimilars 2016. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(3):134-8. doi:10.5639/gabij.2016.0503.034
5. Roundtable on biosimilars with European regulators and medical societies, Brussels, Belgium, 12 January 2016. Generics and Biosimilars Initiative Journal (GaBI Journal). 2016;5(2):74-83. doi:10.5639/gabij. 2016.0502.019
6. European Medicines Agency. European Commission, Biosimilars in the EU. Information guide for healthcare professionals [homepage on the Internet]. [cited 2018 Jul 12]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Leaflet/2017/05/WC500226648.pdf
|
Author for correspondence: Robin Thorpe, PhD, FRCPath, Deputy Editor-in-Chief, GaBI Journal |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2018 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


