What pricing and reimbursement policies to use for off-patent biologicals in Europe? – results from the second EBE biological medicines policy survey
Published on 2017/03/10
Generics and Biosimilars Initiative Journal (GaBI Journal). 2017;6(2):61-78.
Author byline as per print journal: Jean-Baptiste Reiland, MSc; Barbara Freischem; Alexander Roediger, MA
|
Introduction/Study objective Biosimilar policies with a focus on uptake have received a lot of attention at national and regional level in the last few years. It is now 10 years since the first biosimilar was approved in the European Union (EU), but do policies in the Member States take the diff erences between biological medicines and small-molecule generics into account? To map the policy landscape in Europe, European Biopharmaceutical Enterprises (EBE) has conducted a second round of its descriptive survey on pricing and reimbursement policies for off-patent biologicals. |
Submitted: 9 January 2017; Revised: 17 March 2017; Accepted: 19 March 2017; Published online first: 8 May 2017
Introduction/Study objective
Pharmaceutical pricing and reimbursement policies aim to improve the rational use of medicines and keep pharmaceutical spending under control [1]. In the European Union (EU), the design and implementation of pharmaceutical policies are in the competence of Member States as laid down in the Treaty [2]. Decisions about price and reimbursement of prescription medicines are generally made by governments and payers. One particular area of interest for pharmaceutical policy is the off-patent market since with patent expiration, many follow-on medicines, both small-molecule generics and biosimilars, can enter the market and compete with the originator products. For small-molecule medicines, according to Medicines for Europe, governments have saved Euros 100 billion in 2014 thanks to generics [3].
Although biosimilars are not the same as generics [4], they are both marketed to compete with originators [5]. According to IMS Health (now Quintiles IMS) the cumulative potential savings to health systems in the EU and the US, as a result of the use of biosimilars, could exceed Euros 50 billion in aggregate over the next five years (by 2020) and reach as much as Euros 100 billion [6].
Between 2006 and 2016, 23 biosimilars were authorized in the EU, two of which have been withdrawn (2008 and 2012) for commercial reasons. The remaining 21 products only represent 12 biosimilar molecules [7]. Thirteen biosimilars are currently under review by the European Medicines Agency (EMA) [8].
Pharmaceutical policies for the off-patent market for smallmolecule generics traditionally included a variety of measures related to pricing, reimbursement, market entry and expenditure controls but also measures targeting distributors, physicians and patients [1]. Some of the most common policies involve generics substitution, Internal Reference Pricing (IRP) (including Generic Reference Pricing (GRP) and Therapeutic Reference Pricing (TRP)), prescribing by International Nonproprietary Name (INN) of the active principle of the medicine, often linked with the obligation for pharmacists to dispense the cheapest medicine available with the same INN, quotas and finally also procurement practices, such as tendering, see Table 1 in Annexes.

All of these policies aim at creating savings through competition. They have in common that they assume that products are interchangeable, fostering therefore price competition and generation of savings in the off-patent market.
However, biosimilars, with the complex structure of biological medicines, are not the same as small-molecule generics [4], which have simpler chemical structures and are considered to be identical to their reference medicines [9, 10]. EMA Procedural Advice furthermore specifies: ‘The decisions on interchangeability and/or substitution rely on national competent authorities and are outside the remit of EMA’ [11], see Table 1 in Annexes for definitions of interchangeability and substitution used in the context of this paper.
Literature suggests that biosimilar competition is likely to differ from small-molecule generic drug competition: besides the entry hurdles – more intense research and development investment, higher manufacturing costs – Mestre-Ferrandiz et al. point to concerns among some physicians requiring more intense education, and concludes that, ‘… we can expect fewer biosimilar entrants and consequently less intense biosimilar price competition’, [12]. However, other articles also suggest that the intensity of the price competition could be stronger, notably based on the price decreases observed for some biosimilars already marketed for several years in Europe [13].
For instance, some political actors like the Belgian Government considered the uptake of biosimilars as too slow and put in place measures to increase it [14]. Additionally, in 2015 some regulatory agencies, e.g. Finland, Germany, Italy and The Netherlands have issued positions on interchangeability which either allow or encourage switching [15–17]. Nonetheless, none of them does discuss pharmacy-level substitution – Finland and Germany even exclude it from the scope of the position – and all of them have in common that they require physician involvement in treatment changes.
The European Biopharmaceutical Enterprises (EBE) survey on policies for biological medicines intended to examine the current policies and the role of physicians in this context. Acknowledging that design and implementation of pharmaceutical policies are in the competence of Member States, policy measures adopted at national level rarely remain isolated. The objective of EBE in carrying out this survey was to provide a pan-European landscape, showing common trends and differences among countries in the definition of the policies.
Methods
Between February and May 2016, EBE conducted a survey in 32 countries, the 28 EU Member States plus Norway, Serbia, Switzerland and Turkey. This geographical scope, which goes beyond the political borders of the EU, covers countries which are represented within the European Federation of Pharmaceutical Industries and Associations (EFPIA).
The questionnaire was a revised version of the previous survey of 2014 and developed by the EBE Biosimilars Working Group. It contained 44 questions on eight policy areas: Availability of biological medicines, Tendering, Health Technology Assessment (HTA), International Nonproprietary Name (INN) prescribing, Internal Reference Pricing (IRP), Substitution, Interchangeability, and Quotas, see Table 2 in Annexes. The definitions used in the survey and in this paper are shown on Table 1 of Annexes and are based on the Consensus Information Document ‘What you need to know about biosimilar medicinal products’ published by Directorate General Growth, European Commission, in 2013 [18].

The policy areas were selected based on a list of 23 different policies discussed in the Economic Paper 461 ‘Cost-containment policies in public pharmaceutical spending in the EU’ of the Directorate-General for Economic and Financial Affairs (DG ECFIN) [1]. Information for specific countries were based on individual comments provided in the survey response and is used to supplement the findings. The questionnaire was available in English only.
The questionnaire was sent to national pharmaceutical associations, which are members of EFPIA, see Table 5 in Annexes: the initial mailing took place on 4 February 2016 with some reminders, followed by two review mailings on the survey outcome. Participation in the survey was voluntary and did not incur any reimbursement or payments for participation.
The findings presented in the paper are based on the insights provided by the 32 pharmaceutical trade associations surveyed and are not based on a systematic review of the literature.
Results
European Biopharmaceutical Enterprises (EBE) received responses from all 32 national pharmaceutical associations (a 100% response rate).
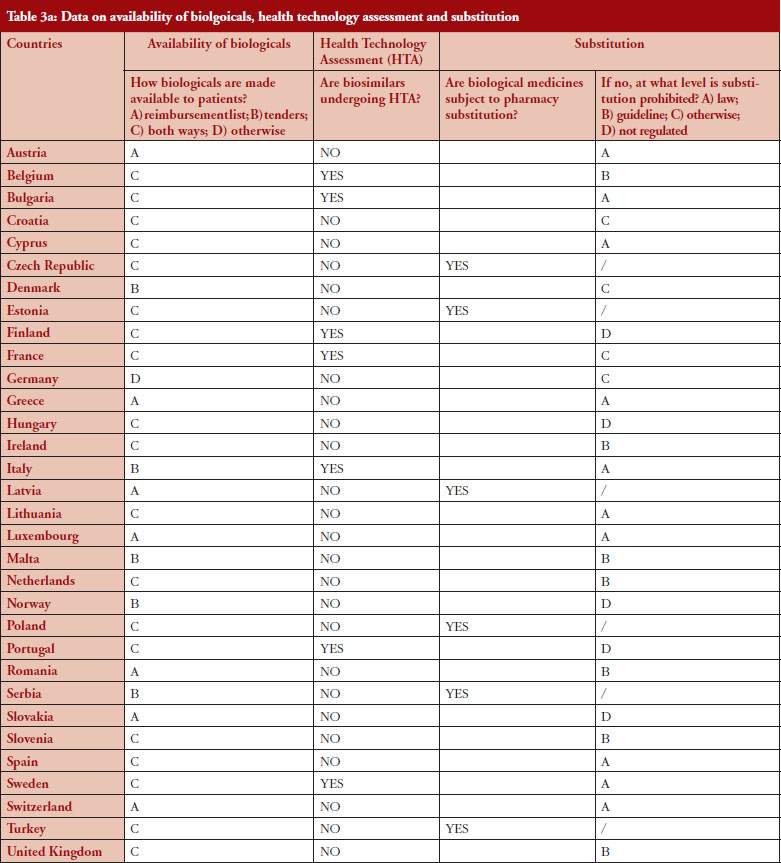
Substitution
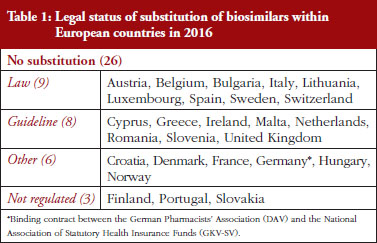
Substitution refers to the practice of dispensing one medicine instead of another equivalent and interchangeable medicine at the pharmacy level without consulting the prescriber, see Table 1 in Annexes. Out of the 32 countries which responded to the survey, 26 (81%) reported that pharmacy-level substitution of biologicals was prohibited. Among these 26 countries, 17 (65%) answered that either laws or guidelines are in place. For the others, answers indicated that prohibition of pharmacylevel substitution is notably linked to the absence of reference to biologicals in the current legal framework or to guidelines by medical societies, e.g. Portugal, or the Regulatory Agency, e.g. Finland, see Table 1.
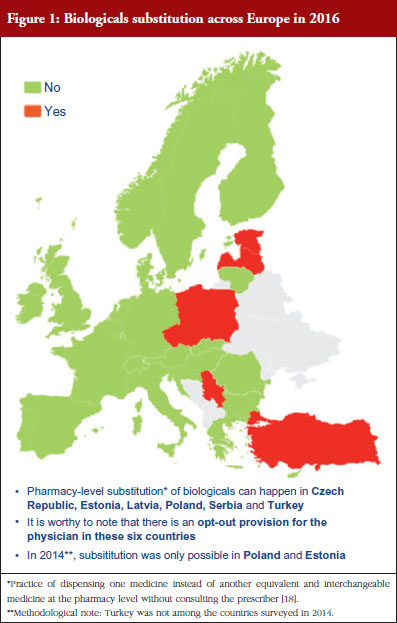
Respondents indicated that pharmacy-level substitution of biologicals could occur in six out of the 32 European countries (19%): Czech Republic, Estonia, Latvia, Poland, Serbia and Turkey, see Figure 1. In 2014, substitution was only possible in Poland and Estonia [19]. It should be noted that these substitution policies are applicable to both ‘naïve’ patients and patients on treatment. Eventually, there are opt-out provisions, which mean that a physician can exclude this possibility of substitution in its prescription form.
Additionally, it should also be mentioned that the answers clearly highlighted that the modalities of pharmacy-level substitution can vary from one country to another. For example, Estonia reported that the possibility of substitution depends on the molecules dispensed in pharmacies while in Czech Republic, due to the fact that there are no special rules for biologicals, pharmacy-level substitution is not prohibited as such. However, it should be recalled that pharmacy-level substitution for biologicals is not automatic and does not occur frequently. In Poland, pharmacy-level substitution is possible, unless the physician indicates ‘Not substitutable’; the pharmacist has to inform about the cheapest option, as there is no differentiation between small molecule drugs and biologicals in the current legislative framework.
In Germany, it should be noted that there is a distinction for biological medicines depending on whether it is considered as a bioidentical or not. Only bioidenticals (= biotechnology drugs with the same active ingredient, i.e. they must not differ as regards their starting materials and manufacturing process) are subject to substitution by the pharmacist. For all other biopharmaceuticals, automatic substitution at pharmacy level is prohibited.
In France, regulation adopted in 2014 allowed, in principle, pharmacy-level substitution at treatment initiation, pending the publication of an implementing decree. Note: When the survey was performed (February–May 2016), this decree had not yet been published and therefore substitution did not take place in practice.
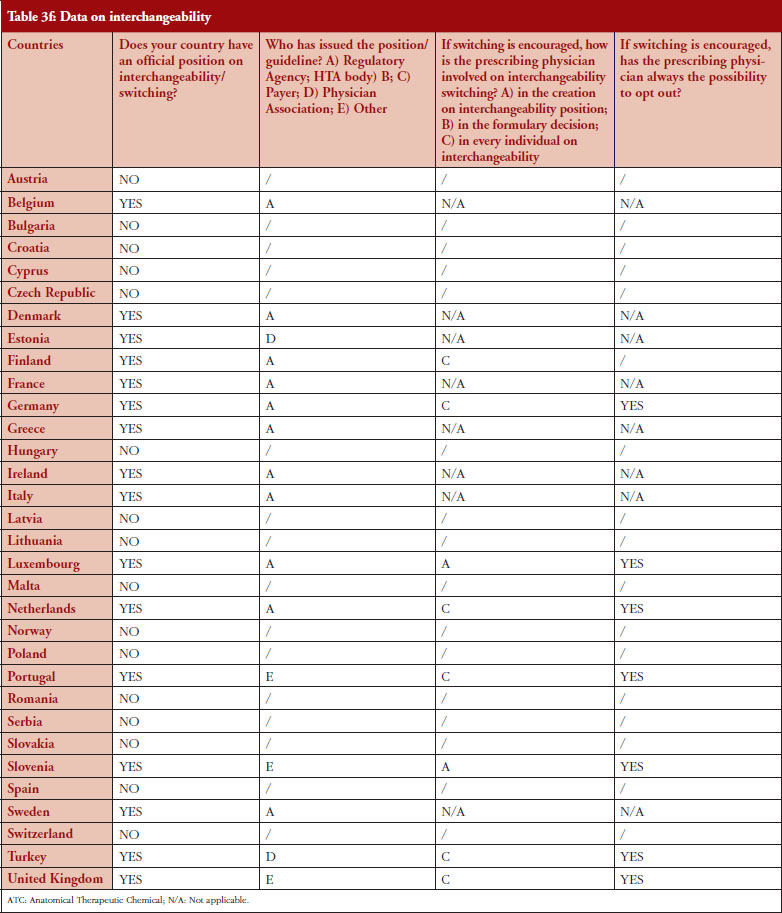
Interchangeability
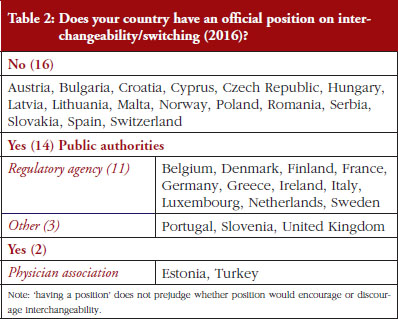
Out of the 32 countries surveyed, 16 (50%) responded that their countries have an official position on interchangeability or switching, see Table 2. It should be mentioned that having a position does not prejudge whether this position would encourage or discourage interchangeability.
Even though the responses indicate that there is no clear and harmonized approach across the European countries on interchangeability, the majority of positions have been issued by the regulatory agency and not by an HTA body or a payer. In 2015, Finland [15] and the Netherlands [17] had been among the first EU countries to adopt a position on interchangeability or switching between a reference product and a biosimilar, allowing physicians to switch under adequate control and surveillance. Germany, through the Paul Ehrlich Institute, adopted a specific position regarding the use of biosimilar infliximab [16].
In 2016, countries like France and Portugal updated or issued guidelines or recommendations. The French agency notably stated that due to the growing body of experience, it is no longer justified to formally exclude all interchangeability. However, patients should be informed, agree to the switch and be closely monitored. Guidelines also state that traceability for concerned products should be ensured [20]. For Portugal, the National Commission on Pharmacy and Therapeutics (Comissão Nacional de Farmácia e Terapêutica, CNFT), a consultative body of the Portuguese regulatory agency under guardianship of the Ministries of Health and Finance, clearly state that the switch between biosimilar medicines must be articulated with the clinical services involved, respecting the precautionary principle and in accordance with the therapeutic indications for each case.
The official positions on interchangeability published across the countries surveyed generally affirm the role of the treating physician as the initiator and supervisor of the switch.
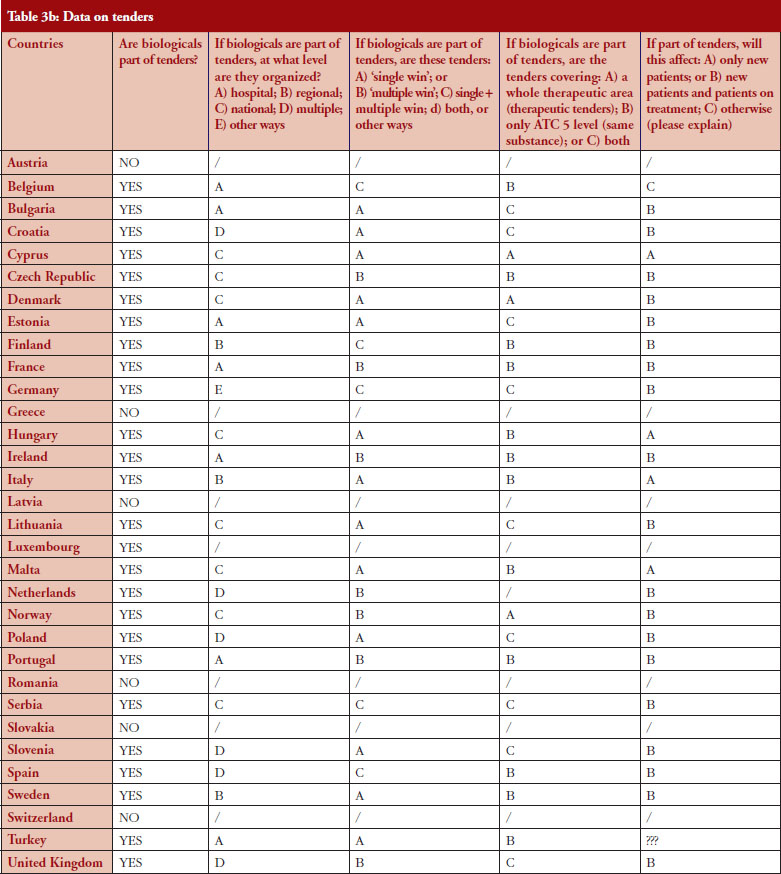
Tendering
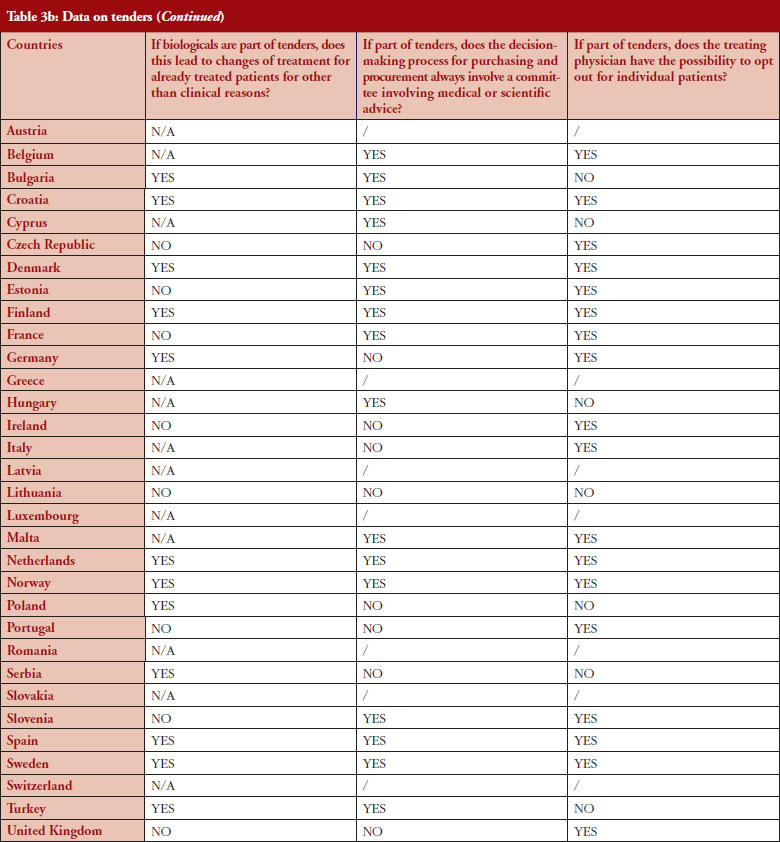
According to the answers received from the trade associations surveyed, biological medicines can be purchased through tendering procedure [21] in 26 out of 32 countries (81%), see Figure 2. Respondents indicated that most of the tenders that include biologicals are either national (8) or hospital tenders (7). Likewise, a large number of countries reported ‘single winner’ tenders (12), which means that only one supplier can win.
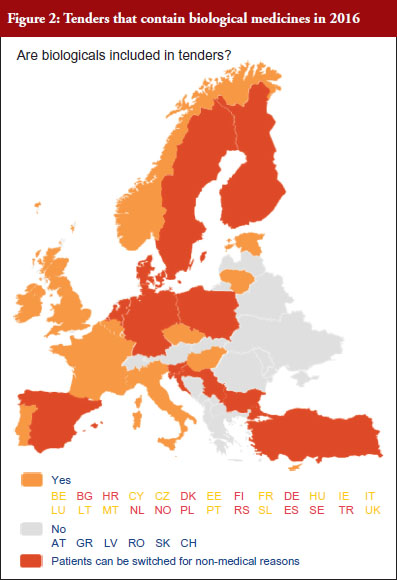
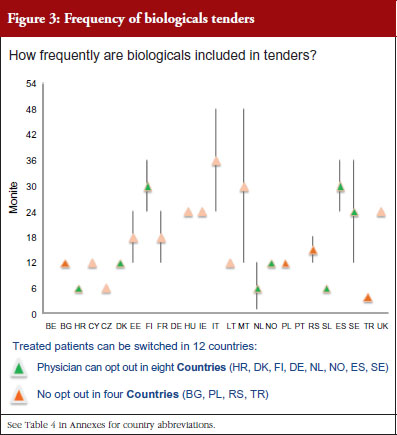
The respondents reported that, as a result of tenders, switching of patients for non-medical reasons can happen in 12 out of the 26 countries (46%), and that in eight of these 12 countries the physician can opt out. This would imply that in four countries patients are forced to switch (Bulgaria, Poland, Serbia and Turkey).
Figure 3 shows how often tenders are conducted, e.g. in Bulgaria every 12 months; in Finland every 24–36 months. Frequency of tendering may have an impact on switching. In Bulgaria, physicians cannot ‘opt out’, which means that they have to prescribe the medicine of the manufacturer which has won the tender.
Health Technology Assessment (HTA)
In the context of the survey, HTA related to the broader assessment of the economic impact of biosimilars. The responses for 75% of the countries indicated that HTA processes do not apply to biosimilars, see Table 3. In the eight countries where biosimilars are reported to undergo HTA, conditions and modalities are not harmonized and vary from one country to another. For example, in Finland, it was reported that biosimilars do not undergo HTA in hospitals but do so in the outpatient care setting while in France, the assessment is conducted through a simplified procedure, with clinical evidence analysed to assess the Therapeutic Value (SMR) and Added Therapeutic Value (ASMR). In Portugal, it was reported that a simplified procedure is in place for biosimilars. There is no assessment of clinical value, only pricing negotiations. Finally, in Hungary it was reported that biosimilars do not undergo HTA except if the product sets a higher price than the reference product.

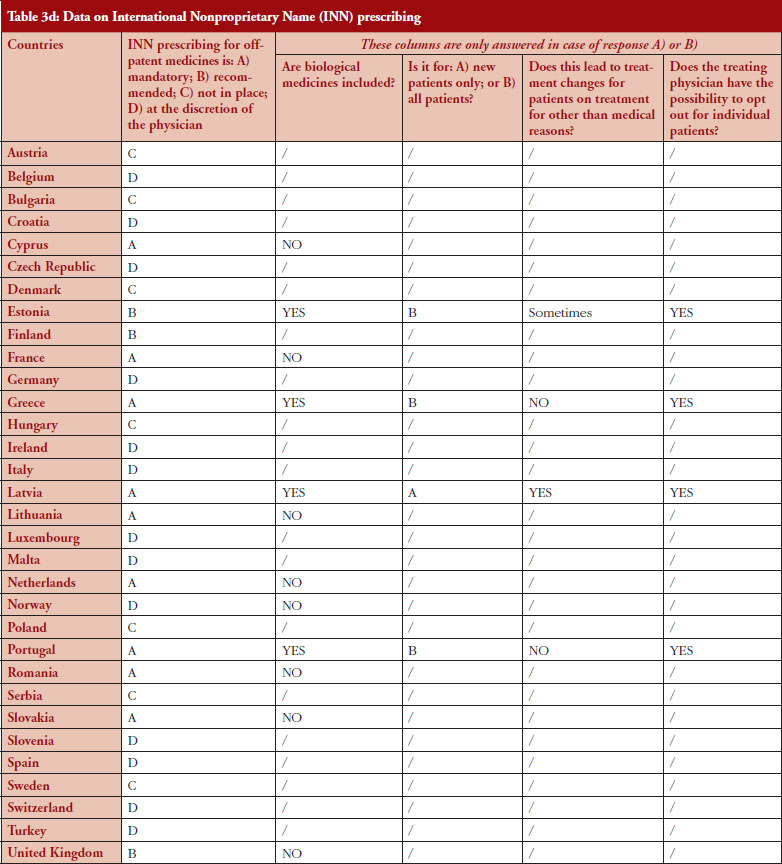
INN prescribing
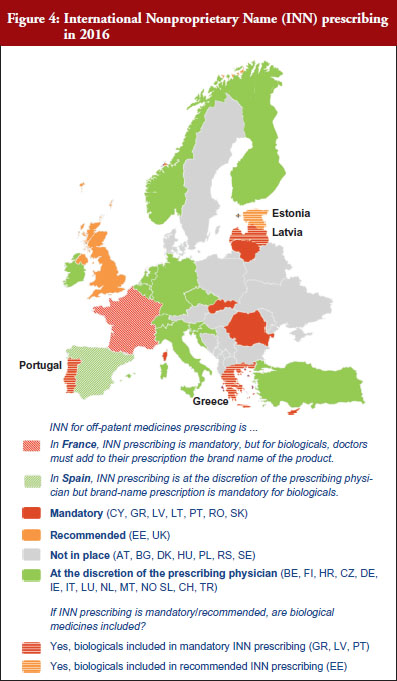
The intention of INN prescribing, meaning prescribing by the INN of the active ingredient of the medicine, is to disable incentives for brand-name prescribing. Prescribing biological medicines by INN can lead to therapeutic changes for patients on treatment since the INN used for the biosimilar is in most cases the same as the one designated by the World Health Organization (WHO) for the reference product.
INN prescribing has been reported as either mandatory or recommended in 11 out of 32 countries (34%). In addition, it appeared from the responses that where INN prescribing was mandatory or recommended, the large majority of countries have introduced mechanisms to exempt biological medicines from prescribing by INN. In seven out of these 11 countries the responses indicated that biologicals are excluded from INN prescribing, which means that although INN prescribing for biologicals is recommended in Estonia, Greece, Latvia and Portugal, effectively, treatment changes may happen only in Estonia and Latvia, see Figure 4.
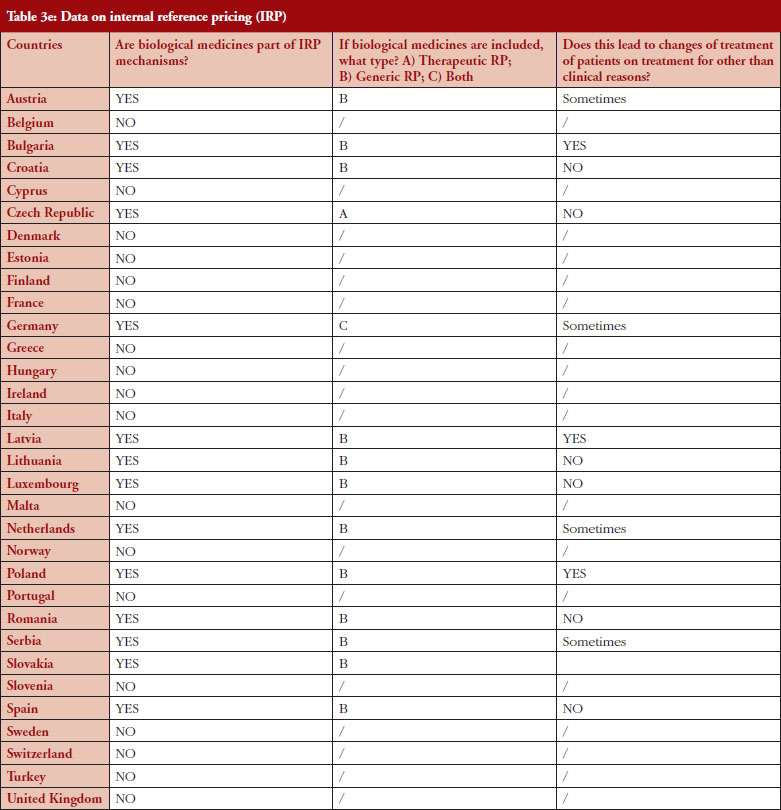
Internal reference pricing (IRP)
For 14 (44%) out of 32 countries the responses indicated that biologicals are included in IRP. Two countries declared applying Therapeutic Reference Pricing (TRP), creating reference groups [22] at the level of the therapeutic class or higher (ATC 4 [Anatomical Therapeutic Chemical] or higher). Twelve countries reported having reference pricing at active substance level (ATC 5; Generic Reference Pricing [GRP]) in place. In Germany, the response highlighted that both TRP and GRP can happen, but not at the same time, see Figure 5. Other countries may have excluded biologicals explicitly from IRP or do not have this policy at all [23]. In three out of 14 countries the responses show that switches can happen due to IRP. In Turkey, the trade association answered that biologicals are part of IRP but this is neither TRP nor GRP. Biologicals and biosimilars have 100% of the reference price. Biosimilar products that do not have a reference price, take the original product’s price as a reference.

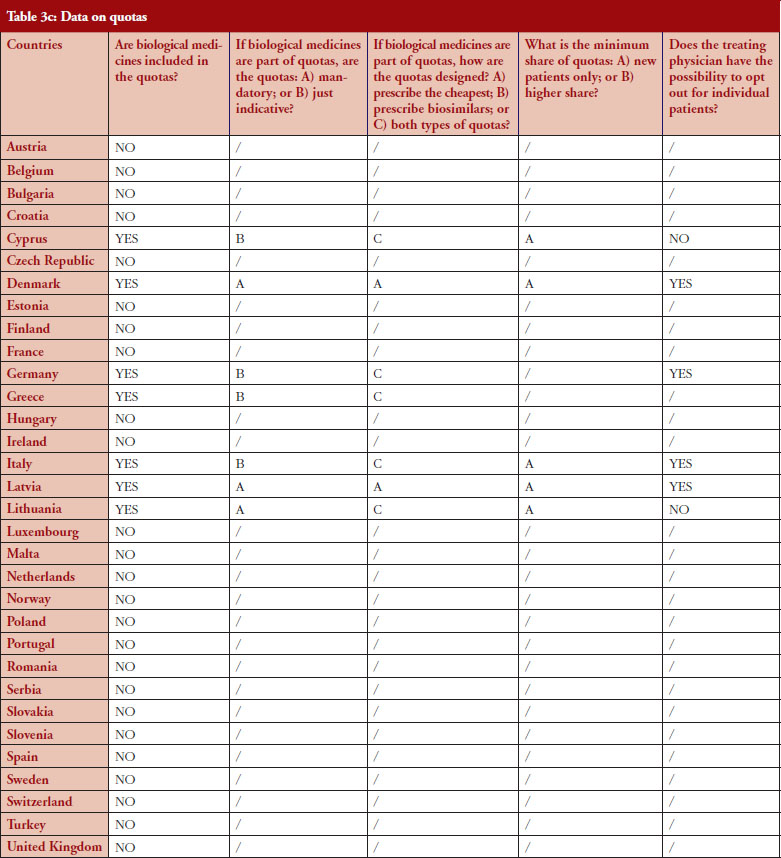
Quotas
According to the consolidated set of responses, quotas of medicine for physicians to increase uptake is not a widely used policy across Europe. Seven countries (22%) have declared having such measures in place: Cyprus, Denmark, Germany, Greece, Italy, Latvia and Lithuania. Nevertheless, none of these countries exclude biologicals from the scope of the quotas, as reported by the respondents, and it appears that there is no ‘one size fits all’ in the design. In three countries (Denmark, Latvia and Lithuania), quotas are mandatory whereas they are only indicative in Germany (except in some regional physicians associations, where physicians are mandated by payers to reach defined quotas), Cyprus, Greece and Italy.
Similar to IRP or INN prescribing, quotas aim to create incentives for prescribing a certain type of medicine. When it comes to the question of whether quotas could create discrimination, i.e. preference for a specific product group, the responses provided highlighted that: in Lithuania, the focus of quotas is on prescribing the cheapest product, whereas in Denmark, the focus of quotas is prescribing biosimilars. In Cyprus, Germany, Italy, Latvia, quotas design is both focused on prescription of the cheapest and on biosimilars. In Portugal, a quota of 20% is established as a performance indicator regarding the National Health Service (Serviço Nacional de Saúde, SNS) funding contract.
The physician’s role
The survey contained several questions on ‘opting out’, i.e. the possibility of the individual prescribing physician not to apply one of the above described policies. This concerns the policy fields of tendering, INN prescribing, substitution, interchangeability and quotas. Opting out is an indicator of the physician’s role when it comes to a treatment decision.
Tendering
Based on the responses of the associations, it appears that optingout is possible in a majority of countries where biologicals are part of tenders (eight out of 12). Out of the 32, 26 countries reported that biologicals are tendered, and 12 countries reported that switching from a reference product to a biosimilar can occur; but physicians can opt out from switching in eight out of the 12 countries even though the degree of freedom to opt out varies between those countries.

INN prescribing of biologicals
Responses show that opting out is always possible even in the four out of 11 countries where INN prescribing for biologicals is mandatory or recommended. However, two countries (Estonia, Latvia) report that the treatment could change due to INN prescribing; nevertheless, the physician has the possibility to opt out.
Substitution
As reported by the respondents, substitution of biologicals at pharmacy level is possible in six countries (Czech Republic, Estonia, Latvia, Poland, Serbia and Turkey). The physician can opt out in all of these countries; however, in some countries it creates some burden on the physician (Estonia, Latvia and Poland).
Interchangeability
Among the 32 countries surveyed, 16 have declared having a position on interchangeability. In six out of eight countries where switching is encouraged, it was reported that the physician decides on every individual switch (Finland, Germany, The Netherlands, Portugal, Turkey and United Kingdom).
Quotas
For seven countries, the existence of quotas for biologicals was reported. The responses also indicate that in four out of these seven countries the physician can opt out (Denmark, Germany, Italy and Latvia); this is not the case in Cyprus and Lithuania. No response on this aspect was received from Greece.
In summary, treatment decisions remain in the hands of physicians in the majority of countries. Based on the complete analysis of the answers provided, it appears that in Latvia and Turkey the physician’s role seems the weakest (tendering, IRP, INN, substitution), followed by Bulgaria, Lithuania, Poland and Serbia.
Limitations
The survey had several limitations. It only included questions about biological medicines and did not poll generics policies in the respective countries.
Organization of healthcare systems, including pricing and reimbursement of medicines, is a national competence in Europe, and it is acknowledged that any comparison of policy frameworks is limited by the need to streamline questions versus the desire to capture the subtleties of the various systems.
The questions, see Table 1 in Annexes, were general by nature and were thus not able to probe the details and interdependencies around those policies. However, many respondents added country specific comments, thus providing better granularity for the analysis. To minimize the risk of inappropriate comparisons, only data which allowed solid comparisons are presented. Certain data such as opt-out for physicians in IRP were therefore excluded from the analysis since the diversity of the health systems precluded meaningful conclusions.
The findings presented in the paper are based on the insights provided by the 32 pharmaceutical trade associations surveyed. There was no validation process with experts or through literature review.
Discussion
The results of this second EBE policy survey show that, considering the increased entry of biologicals and biosimilars in the health systems, a large majority of the countries in Europe have specific policies in place reflecting the different nature of biological medicines compared with small-molecule medicines.
It appears from the responses of the trade associations surveyed that the prescribing physician retains a decision-making role. In nearly all of the countries and policy areas the treatment decision remains with the physician. This is in line with the Policy Principles for Off-patent Biologic Medicines in Europe of EFPIA [24].
Mestre-Ferrandiz et al. have recently assessed different policies and their contribution to ‘achieve significant, sustained savings from use of biosimilars’: 1) substitution; 2) direct price intervention, e.g. IRP or price cuts; 3) tendering; 4) ‘incentives for budget holders to use lower-cost products’; and 5) market support through outcomes data for physicians and pharmacists [12]. Drawing on the experiences from the generics market, the authors suggest that policy measures 4) and 5) would be appropriate policies to ensure long-term savings from biosimilars competition. Other literature by Curto et al. suggest that tendering (which would therefore be the measure 3 of Mestre-Ferrandiz et al.) might be the most efficient way of fostering competition and generating ‘worthwhile savings can be generated in tenders, once the bid is designed in such a way that competition can produce its effects, i.e. allowing more than one manufacturer to tender’ [25].
The results from the present EBE biologicals policy survey show that the majority of countries are following this approach. Most countries prohibit substitution at pharmacy level, and the majority of countries, which practice INN prescribing and IRP, exclude biologicals. In addition, physicians can opt out in all six countries where substitution or INN prescribing is in place. Concerning prescribing practices, it is worth mentioning that, further to the adoption of the cross-border healthcare EU Directive [26], an Implementing Directive is requiring the use of brand name when prescribing biological medicines in order to ensure traceability and patient safety [27].
Tendering is used widely. For the vast majority of countries (81%) it was reported that biologicals are included in tenders and that in 50% of these, switching of patients for non-medical reasons can happen, i.e. Bulgaria, Poland, Serbia and Turkey, without opt-out possibility for the prescriber. This can be of concern when also taking into account the frequency of tenders reported in Turkey (every four months), Bulgaria and Poland (every 12 months each), as it could lead to multiple treatment changes all of which are out of the prescribing physician’s control.
Tenders can generate substantial savings if sufficient manufacturers participate in them. However, ‘winner takes all’ tenders can also pose a barrier to entry of competing products and lead to a risk of shortages [12]. In its policy principles EFPIA recommends that when incentives for using lower-cost products are used to increase uptake, they should: 1) explicitly retain doctors’ freedom to prescribe; 2) create a clear level playing field between off-patent originators and biosimilars; 3) propose a level of volume shift in the market that is, at a maximum, in line with a policy of naïve patient initiation; and 4) ensure that treatment decisions are always made first on the basis of clinical judgement and secondly on the basis of an overall value proposition offered by the individual medicine [24].
Regarding the use of quotas, the survey shows a mixed picture with regard to the recommendations of EFPIA principles. Quotas can be used to increase uptake of lower-cost products. Of the countries which declared having quotas in place, some focus on prescribing the cheapest, while others require prescribing a certain amount of biosimilars. Also, some countries fail to retain the physician’s freedom to prescribe.
Uptake is not necessarily an indicator for sustainable savings. The recent IMS study on the impact of biosimilar competition shows that the correlation between biosimilars market share, i.e. uptake, and price was weak [8]. In fact, it was competition between multiple suppliers, off-patent originators and various biosimilars manufacturers which drove down prices. This was not only the case in the market of biosimilars and its respective reference product, but also in the total market.
Since the last EBE biologicals policy survey much has happened in the area of market support, especially in the assessment and monitoring of switching. Several manufacturers, as well as other stakeholders, have initiated switching studies [28]. An overview is also given by Braun et al. [29]. The new and updated positions of the Regulatory Agencies of Finland, Germany and The Netherlands reflect the increasing number of available biosimilars and the need of first assessments through the national regulatory bodies.
Conclusion
The results of this second EBE policy survey show that, considering the increased entry of biologicals and biosimilars in the health systems, a large majority of the countries in Europe have specific policies in place reflecting the different nature of biological medicines compared with small-molecule medicines. In addition, treatment decisions remain in the hands of physicians in the majority of countries, where physicians can either decide on treatments or opt out. However, policies and their implementation vary among different jurisdictions. Compared to the first EBE survey, shifts have been seen in the area of substitution and interchangeability.
Competing interests: This paper is authored by European Biopharmaceutical Enterprises (EBE) and represents the views of the organization.
EBE is an industry association funded by contributions from its membership. EBE’s membership consists of companies of all sizes, ranging from SMEs (Small Medium Enterprises) to big multinationals, as well as organizations supporting the development of biopharmaceuticals. For more information, visit www.ebe-biopharma.eu/
Provenance and peer review: Not commissioned; externally peer reviewed.
Contributing authors from European Biopharmaceutical Enterprises (EBE)
Jean-Baptiste Reiland, MSc, Project Manager, Global Public Affairs, Sanofi
Barbara Freischem, Executive Director, European Biopharmaceutical Enterprises
Alexander Roediger, MA, Executive MBA, Policy Lead Oncology Europe, Middle East, Africa and Canada (EMEAC), MSD
References
1. European Commission. Carone G, Schwierz C, Xavier A. Cost-containment policies in public pharmaceutical spending in the EU. Economic Papers 461. 2012 [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://ec.europa.eu/economy_finance/publications/economic_paper/2012/pdf/ecp_461_en.pdf
2. European Union. Eur-Lex. Treaty of the Functioning of the European Union, Art. 168(7); Official Journal (9.5.2008) C 115/47-199 [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:12012E/TXT
3. Medicines for Europe (2016), The role of generic medicines in sustaining healthcare systems: A European perspective. [cited 2017 Mar 17]. Available from: http://www.medicinesforeurope.com/wp-content/uploads/2016/03/infographicims.pdf
4. European Commission. What I need to know about biosimilar medicines – information for patients. January 2017 [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://ec.europa.eu/growth/tools-databases/newsroom/cf/itemdetail.cfm?item_id=9066.
5. European Medicines Agency. Questions and answers on biosimilar medicines (similar biological medicinal products). EMA/837805/2011. 27 September 2012 [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2009/12/WC500020062.pdf
6. IMS Institute for Healthcare Informatics. Delivering on the potential of biosimilar medicines. The role of functioning biosimilar markets. March 2016 [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://www.imshealth.com/files/web/IMSH%20Institute/Healthcare%20Briefs/Documents/IMS_Institute_Biosimilar_Brief_March_2016.pdf
7. European Medicines Agency. European public assessment reports [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d125
8. European Commission. The impact on biosimilar competition on price, volume and market share – updated version 2016 [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://ec.europa.eu/growth/toolsdatabases/newsroom/cf/itemdetail.cfm?item_id=8854&lang=en
9. Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK, et al. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191-6.
10. European Medicines Agency. Guideline on similar biological medicinal products. CHMP/437/04 Rev 1. 23 October 2014 [homepage on the Internet]. [2017 Mar 17]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf
11. European Medicines Agency. European Medicines Agency procedural advice for users of the centralised procedure for similar biological medicinal products applications. EMA/940451/2011. December 2015 [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2012/04/WC500125166.pdf
12. Mestre-Ferrandiz J, Towse A, Berdud M, Biosimilars: How can payers get long-term savings? Pharmacoeconomics. 2016;34(6):609-16.
13. Garattini L, Curto A, van de Vooren K. Western European markets for biosimilar and generic drugs: worth differentiating. Eur J Health Econ. 2015;16(7):683.
14. De Block M. Biosimilaires: la Belgique doit combler son retard. 7 Janvier 2016 [home page on the Internet]. [cited 2017 Mar 17]. Available from: http://www.deblock.belgium.be/fr/%C2%AB-biosimilaires-la-belgique-doit-comblerson-retard-%C2%BB
15. FIMEA (2015), Interchangeability of Biosimilars – Position of Finnish Medicines Agency Fimea. [cited 2017 Mar 17]. Available from: https://www.fimea.fi/documents/542809/838272/29197_Biosimilaarien_vaihtokelpoisuus_EN.pdf
16. Paul Ehrlich Institut. Position des Paul-Ehrlich-Instituts zur Interchangeability von Biosimilars [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://www.pei.de/EN/medicinal-products/antibodies-immunoglobulins-fusion-proteins/monoclonal-antibodies/biosimilars/position-peiinterchangebility-biosimilars-content.html
17. CBG-MEB. Biosimilar medicines [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://english.cbg-meb.nl/human/for-healthcare-providers/contents/biosimilar-medicines
18. European Commission. What you need to know about biosimilar medicinal products. A consensus information document [homepage on the Internet]. [cited 2017 Mar 17]. Available from: ec.europa.eu/DocsRoom/documents/8242/attachments/1/translations/en/renditions/native+&cd=1&hl=en&ct=clnk&gl=us
19. European Biopharmaceutical Enterprises. What pricing and reimbursement policies to use for off-patent biologicals? – Results from the EBE 2014 biological medicines policy survey. Generics and Biosimilars Initiative Journal (GaBI Journal). 2015;4(1):17-24. doi:10.5639/gabij.2015.0401.006
20. L’Agence nationale de sécurité du médicament et des produits de santé. L’ANSM publie une mise au point sur les médicaments biosimilaires – Point d’Information. 3 Mai 2016 [homepage on the Internet] [cited 2017 Mar 17]. Available from: http://ansm.sante.fr/S-informer/Points-d-information-Pointsd-information/L-ANSM-publie-une-mise-au-point-sur-les-medicamentsbiosimilaires-Point-d-Information
21. European Biopharmaceutical Enterprises. EBE Position Paper on tendering of biologicals, including biosimilars. 21 May 2012 [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://www.ebe-biopharma.eu/documents/3/22/EBE-Position-Paper-on-Tendering-of-Biologicals-including-Biosimilars.
22. Gesundheit Österreich GmbH (GOEG), WHO Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies (2015), Glossary; [cited 2017 Mar 17]. Available from: http://whocc.goeg.at/.
23. Dylst P, Simoens S, Arnold G Vulto. Reference pricing systems in Europe: characteristics and consequences. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(3-4):127-31. doi:10.5639/gabij.2012.0103-4.028
24. European Federation of Pharmaceutical Industries and Associations. EFPIA launches robust principles to secure a competitive European off-patent biologic medicines market [homepage on the Internet]. [cited 2017 Mar 17]. Available from: https://www.efpia.eu/news-events/the-efpia-view/statements-press-releases/150930-efpia-launches-robust-principles-to-secure-a-competitive-european-off-patent-biologic-medicines-market/
25. Curto S, Ghislandi S, van de Vooren K, Duranti S, Garratini L. Regional tenders on biosimilars in Italy: an empirical analysis of awarded prices. Health Policy. 2014;116(2-3):182-7.
26. European Union. Directive 2011/24/EU of the European Parliament and the Council of 9 March 2011 on the application of patients’ rights in cross-border healthcare [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2011:088:0045:0065:en:PDF
27. European Commission. Commission Implementing Directive 2012/52/EU of 20 December 2012 laying down measures to facilitate the recognition of medical prescriptions issued in another Member State [homepage on the Internet]. [cited 2017 Mar 17]. Available from: http://ec.europa.eu/health//sites/health/files/cross_border_care/docs/impl_directive_presciptions_2012_en.pdf
28. EU Clinical Trials Register. Clinical trials for eudract_number:2014-002056-40 [homepage on the Internet]. [cited 2017 Mar 17]. Available from: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2014-002056-40]
29. Braun J, Kudrin A. Switching to biosimilar infliximab (CT-P13): evidence of clinical safety, effectiveness and impact on public health. Biologicals. 2016; 44(4):257-66.
|
Author for correspondence: Jean-Baptiste Reiland, MSc, Project Manager, Global Public Affairs, Sanofi, 54 rue la Boétie, FR-75008 Paris, France |
Disclosure of Conflict of Interest Statement is available upon request.
Copyright © 2017 Pro Pharma Communications International
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.


