The potential of generics policies: more room for exploitation–PPRI Conference Report
Author byline as per print journal: Sabine Vogler, PhD; Nina Zimmermann, MA
|
Introduction: This Conference Report aims to provide an overview of key results, messages and conclusions of the Pharmaceutical Pricing and Reimbursement Information (PPRI) Conference with regard to generics. |
Submitted: 22 May 2012; Revised: 5 August 2012; Accepted: 10 August 2012; Published online first: 31 August 2012
Introduction
On 29 and 30 September 2011, the World Health Organization (WHO) Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies organized the second PPRI Conference in Vienna, Austria. This was attended by 275 delegates (officials and staff of public authorities and payers, pharmaceutical companies, researchers) from 56 countries, and included 60 speakers, panelists and chairs. It addressed pharmaceutical policies from a public health perspective, from both a European and global context.
The first PPRI Conference took place in June 2007 in order to present preliminary results of the research project ‘Pharmaceutical Pricing and Reimbursement Information (PPRI)’ co-funded by the European Commission, Directorate General Public Health and Consumers (DG SANCO) which ran from September 2005 until the beginning of 2008 [1–3]. The first PPRI Conference focused on European pricing and reimbursement policies [4], but generics policies were not a key topic at the conference. Just one presentation, by Professor Richard Laing from WHO, about availability, affordability and price components of medicines in developing and transitional countries [5], highlighting price differences between originators and generic medicines assessed according to WHO/HAI (Health Action International) methodology [6, 7], addressed the generics policy.
The second PPRI Conference was organized in response to requests from participants at the previous conference for a follow-up, and was an activity under the terms of reference of the WHO Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies (established in June 2011). The conference objective was to provide information about the activities and the results of the PPRI and Pharmaceutical Health Information System (PHIS) projects. The PPRI initiative has continued as a voluntary networking and information sharing initiative of competent authorities for pricing and reimbursement after the PPRI project officially ended in 2008. The PHIS project (September 2008 to April 2011) [8] was based on the lessons learned and looked particularly at medicines management in the inpatient sector, for which scant literature existed. As a result, the PHIS project called for an urgent improvement in cooperation at the interface between outpatient and hospital sectors [9].
The second PPRI Conference therefore had a focus on ‘hospital pharma and interface management’ and contained three parallel strands: (1) pricing and reimbursement, (2) rational use of medicines and (3) hospital pharma and interface management. The majority of the participants (more than 50 per cent) attended the strand on ‘pricing and reimbursement’. The topics of generics and generics policies appeared in all three strands.
Panelists and speakers in the plenary sessions
Generics were mentioned in 48 (64%) of the total of 75 contributions (presentations, posters, panel discussions), and in 22 of 48 accepted abstracts (46%). During the first high-level panel discussion Mr Kees de Joncheere, then Regional Adviser for Health Technology and Pharmaceuticals for WHO Europe (now Head of the Essential Medicines Department in WHO Headquarters), stressed the great potential of generics policies and the need to apply these more frequently. Mr Richard Bergstrom, President of the European Federation of Pharmaceutical Industries and Associations stated that ‘once patents expire, prices should fall to a low, but sustainable, level’. In this panel discussion but also throughout the conference there appeared to be a common understanding that generics competition works well. Dr Andreas Seiter, Senior Health Specialist, World Bank [10], expressed concern about having a pricing regulation policy that fixes a difference between the originator and the generic(s) (so-called ‘generic price link’ [11]) because this could impede further benefits and savings that could be achieved through full generics competition.
Country case studies
Presentations and posters described the pharmaceutical situation in 37 different countries, including generics policies and their outcomes. In Croatia, for instance, a generic price link and reference price system, together with policy measures targeting new medicines, have contributed significant savings to the Croatian Social Insurance in 2009 and 2010. Expenditure by Croatian Social Insurance on medicines in the outpatient sector corresponded to Euros 393 million in 2010, 2.9% lower than in the previous year. And in the first months of 2010, public pharmaceutical expenditure in the outpatient sector was 13% lower than that of the first half year of 2009, resulting in a 22% reduction in the health insurance deficit. The savings enabled 47 new innovative medicines to be added to the positive list, i.e. the list of medicines that may be prescribed at the expense of a public payer [12] and 13 medicines to the list of expensive hospital products from July 2009 to July 2010 [13].
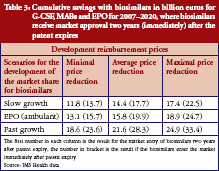
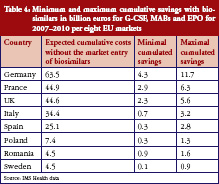
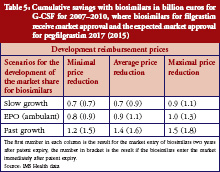
Generics policies were highlighted as a policy option for countries, e.g. Ireland, Portugal, and Spain, that needed to implement a bundle of measures in order to respond to the global financial crisis. The crisis created, as one participant [14] commented, ‘a sense of urgency’ to implementing measures. Dr Miguel Gomes from the Portuguese Medicines Agency [15] reported that following the ‘Memorandum of Understanding’ with the ‘Troika’ (International Monetary Fund, European Central Bank, European Commission), savings measures were swiftly implemented. Previously, Portugal’s pricing system for generics set a reference price, i.e. reimbursement amount within a cluster of medicines of identical active ingredient, dosage and pharmaceutical form [12]; defined by the highest priced generics in the cluster [11, 16]. This situation had been criticized as a lost opportunity for savings [17, 18]. Portugal was the only country of the EU to have higher generics market shares in value than in volume [1], which is an indicator of a high price level of generic medicines. A change in methodology in 2010 led to the reference price being redefined as the average of the five cheapest generics [19]. Dr Gomes presented data on how the generics market had developed well, from a starting point of 3.5% in volume in 2003 to a break-even point in 2010 at which generics shares became lower in value than in volume, see Figure 1. Additional data on the development of the average generics prices confirmed a reduction in prices of generic medicines from the first months of 2010 onwards [15]. Generics policies therefore need to be designed for maximum potential benefit.
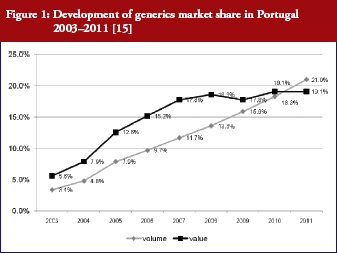 [15]
[15]
Case studies of enhancing generics uptake
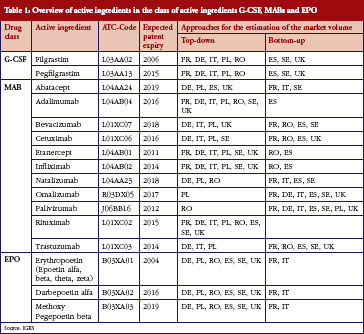
Some European countries reported about differences in price between original products and generic medicines, for example, regarding generic omeprazole and simvastatin [20], see also the article [21] generic ACE (angiotensin-converting-enzyme) inhibitors and SSRI’s (selective serotonin re-uptake inhibitors). Dr Kristina Garuoliene, from the Lithuanian National Health Insurance Fund demonstrated how her country, with its small population, was able to achieve considerable price reductions for generic versus originator medicines, see Table 1 [22]. Dr Kristina Garuoliene also presented a case study of clopidogrel and described how public authorities and payers were concerned about how differences in the salt composition, and indications, between originator and generic clopidogrel could potentially reduce savings from generics availability. Responding to efforts by the originator company to retain sales, authorities adopted ‘pragmatic approaches’ to enhance generics prescribing, e.g. mandatory generics substitution, educational activities, see Table 2 [23, 24]. Still, the differences in the reimbursed prices of generic clopidogrel versus the originator were high across Europe, especially briefly after launch, but decreasing over time.
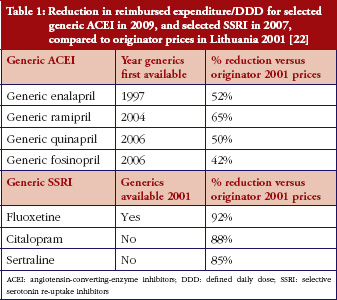 [22]
[22]
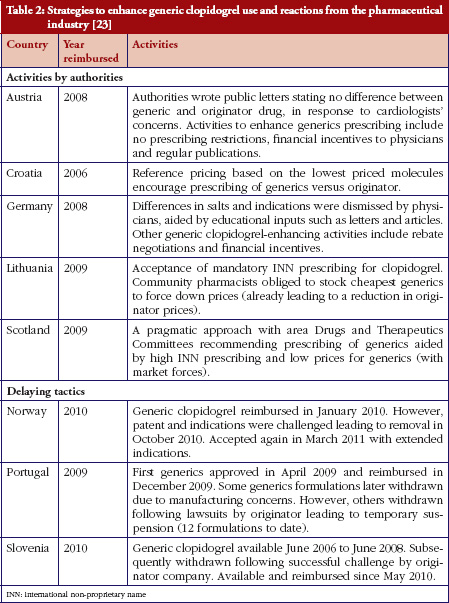 [23]
[23]
In Abu Dhabi, a new policy for generic medicines, including compulsory international non-proprietary name prescribing, was introduced in 2009. However, the expected savings did not fully materialize since there were no accompanying measures to support an increased generics uptake. Abu Dhabi is now exploring a policy of introducing demand-side measures, including educational activities and the introduction of a reference price system for medicines grouped together if they had the same active ingredients [25].
GaBI (Generics and Biosimilars Initiative) at the ‘Meet the Editors’ session
The relevance of generics was highlighted at the fringe ‘Meet the Editors’ session where Dr Brian Godman presented the Generics and Biosimilars Initiative Journal (www.gabi-journal.net), of which he is a member of the International Editorial Advisory Board.
Conclusion
In the final panel discussion, experts from the Norwegian Directorate of Health, OECD, the Southern African Development Community, the WHO Collaborating Centre at Harvard University and WHO Europe stressed the importance of generics policies. The following consensus statement was issued in a conclusion document: ‘Generic[s] policies appear not to be fully exploited yet. At the PPRI Conference, generics were identified as one area where competition works. There is common understanding that savings from generics might be invested for funding innovation. However, as evidence on generics penetration across the countries demonstrated, generics uptake could be improved by more consistent generic[s] policies’ [26]. The PPRI Conference confirmed that in order to achieve the best possible benefits from generics policies, they have to be carefully designed and implemented in a consistent way.
This is an original report from the PPRI Conference. Conference presentations, abstracts and posters are publicly available from: http://whocc.goeg.at/Conference2011/Programme
For patients
The PPRI Conference offered several good practice examples as well as a few less successful stories about the implementation of generics policies. Such sharing of information is crucial for policymakers to know the impact of generics policies. Well-informed policymakers can contribute more effectively towards improving the accessibility of medicines, e.g. by applying generics policies in a consistent way and thus creating opportunities for innovation.
A further conclusion from the conference was that the perspectives of consumers and patients should be taken into account. Participants were reminded that pharmaceutical policies should benefit all citizens, especially vulnerable groups, and that their perspectives should be actively considered.
Acknowledgements
We are very thankful to all who contributed to the success of the PPRI Conference. First of all, we thank the scientific programme committee who supported the organizers in the development of the agenda including the selection of abstracts. Members of the scientific programme committee of the PPRI Conference included Mr Jaime Espin Balbino from the Andalusian School of Public Health (EASP); Ms Margaret Ewen from Health Action International (HAI); Mr Kees de Joncheere, WHO; Professor Richard Laing, WHO; Ms Aukje Mantel-Teeuwisse, Utrecht University, WHO Collaborating Centre for Pharmacoepidemiology and Pharmaceutical Policy Analysis; Mr Øyvind Melien, Norwegian Directorate of Health; Ms Claudia Habl from the Austrian Health Institute (GÖG); and the authors of this manuscript. Further, we thank all speakers, panelists and chairs for their contributions. We particularly thank all those who submitted a large number of high quality abstracts, and those who actively participated with questions and comments. Finally, we are grateful to colleagues in the Austrian Health Institute for their organisational help.
Sources of support
The PPRI Conference was organized by the WHO Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies, located at the Health Economics Department of Gesundheit Österreich GmbH/Geschäftsbereich ÖBIG – Austrian Health Institute under its Terms of Reference as a WHO Collaborating Centre (//apps.who.int/whocc/Detail.aspx?cc_ref=AUT-14&cc_city=vienna&). Funding for the Vienna WHO Collaborating Centre’s activities is provided for by the Austrian Federal Ministry of Health who is the legal owner of the Austrian Health Institute. The PPRI Conference was open to anybody interested in the field. Participants were charged a conference fee to cover logistical and organizational costs.
Competing interests: None.
Provenance and peer review: Commissioned; externally peer reviewed.
Co-author
Nina Zimmermann, MA, Researcher, Geschäftsbereich ÖBIG, Austria
References
1. Vogler S, Habl C, Leopold C, Rosian-Schikuta I. PPRI Report. Vienna: Gesundheit Österreich GmbH/Geschäftsbereich ÖBIG; 2008.
2. Vogler S, Espin J, Habl C. Pharmaceutical Pricing and Reimbursement Information (PPRI) – New PPRI analysis including Spain. Pharmaceuticals Policy and Law. 2009;11(3):213-34.
3. Arts D, Habl C, Rosian I, Vogler S. Pharmaceutical Pricing and Reimbursement Information (PPRI): a European Union project. Italian Journal of Public Health. 2006;3(1):36-40.
4. Vogler S, Habl C, Leopold C, Rosian-Schikuta I. Agenda. PPRI Conference; Vienna: PPRI (Pharmaceutical Pricing and Reimbursement Information); 2007.
5. Laing R. Pharmaceutical Pricing – Availability, Affordability and Price Components in developing and transitional countries. PPRI Conference; Vienna: PPRI (Pharmaceutical Pricing and Reimbursement Information); 2007. Available from: http://ppri.goeg.at/Downloads/Presentations/05_WHO_Geneve_Mr_Laing.pdf
6. Gelders S, Ewen M, Nogucchi N, Laing R. Price, availability and affordability. An international comparison of chronic disease medicines. Background report prepared for the WHO Planning Meeting on the Global Initiative for Treatment of Chronic Diseases, Cairo, December 2005. Buch, Monographie. Cairo: World Health Organization, Regional Office for the Eastern Mediterranean, 2006.
7. WHO, Health Action International (HAI). Medicine prices: a new approach to measurement. Working draft for field testing and revision. 2003.
8. Hoebert J, Mantel-Teuwisse A. PHIS (Pharmaceutical Health Information System) Evaluation Report. Utrecht: Utrecht WHO Collaborating Centre for Pharmacoepidemiology and Pharmaceutical Policy, 2011.
9. Vogler S, Habl C, Leopold C, Mazag J, Morak S, Zimmermann N. PHIS Hospital Pharma Report. Vienna: Pharmaceutical Health Information System; Gesundheit Österreich GmbH / Geschäftsbereich ÖBIG; 2010.
10. Seiter A. Pricing and reimbursement of medicines from global perspective. PPRI Conference. Vienna: PPRI (Pharmaceutical Pricing and Reimbursement Information); 2011. Available from: http://whocc.goeg.at/Downloads/Conference2011/PraesentationenPPRIKonferenz/Day2_afternoon_Festsaal_1400_Seiter.pdf
11. Vogler S. The impact of pharmaceutical pricing and reimbursement policies on generics uptake: implementation of policy options on generics in 29 European countries – an overview. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(2):93-100. doi:10.5639/gabij.2012.0102.020
12. WHO Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies [homepage on the Internet]. Glossary of pharmaceutical terms. Vienna: 2011 [latest update of print version: 2011; regularly updated online; cited 2012 Aug 31]. Available from: http://whocc.goeg.at/Glossary/Search
13. Voncina L, Strizrep T, Godman B, Vlahovi-Palevski V. Recent policies to reduce drug costs and the budget deficit in Croatia: impact and example to others. PPRI Conference Abstract Book; Vienna: PPRI (Pharmaceutical Pricing and Reimbursement Information); 2011. Available from: http://whocc.goeg.at/Downloads/Conference2011/Abstract_poster_book.pdf
14. Koopmanschap M. Generic medicine pricing: on track in Europe? Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(1):5. doi:10.5639/gabij.2012.0101.002
15. Gomes M. Pricing and reimbursement policies in the light of the financial crisis. Country examples: Portugal. PPRI Conference. Vienna: PPRI (Pharmaceutical Pricing and Reimbursement Information); 2011. Available from: http://whocc.goeg.at/Downloads/Conference2011/PraesentationenPPRIKonferenz/Day1_afternoon_Festsaal_1300_Gomes.pdf
16. Teixeira I, Vieira I. PPRI Pharma Profile. Portugal. Vienna: PPRI (Pharmaceutical Pricing and Reimbursement Information); 2008.
17. Habl C, Vogler S, Leopold C, Schmickl B, Fröschl B. Referenzpreissysteme in Europa. Analyse und Umsetzungsvoraussetzungen für Österreich. Wien: ÖBIG Forschungs- und Planungsgesellschaft mbH; 2008.
18. Vogler S, Leopold C. Access to essential medicines in Portugal. Vienna: ÖBIG Forschungs- und Planungsgesellschaft mbH; 2009.
19. Vogler S, Zimmermann N, Leopold C, de Joncheere K. Pharmaceutical policies in European countries in response to the global financial crisis. Southern Med Review. 2011;4(2):22-32.
20. Godman B, Burkhardt T, Garuoliene K, Teixeira I, Tulunay FC, Gustafsson L. Trends in generic pricing policies in Europe: implications for sustaining equitable and comprehensive healthcare. PPRI Conference Abstract Book; Vienna: PPRI (Pharmaceutical Pricing and Reimbursement Information); 2011. Available from: http://whocc.goeg.at/Downloads/Conference2011/Abstract_poster_book.pdf
21. Godman B, Wettermark B, Bishop I. European payer initiatives to reduce prescribing costs through use of generics. Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(1):22-7. doi:10.5639/gabij.2012.0101.007
22. Garuoliene K, Gulbinovi J, Godman B, Wettermark B, Haycox A. European countries with small populations cannot obtain appreciable price reductions for generics: Lithuania – case history to contradict this. PPRI Conference Abstract Book; Vienna: PPRI (Pharmaceutical Pricing and Reimbursement Information); 2011. Available from: http://whocc.goeg.at/Downloads/Conference2011/Abstract_poster_book.pdf
23. Baumgärtel C, Garuoliene K. Enhancing the utilisation of generic clopidogrel: a case history for future guidance? PPRI Conference. Vienna: PPRI (Pharmaceutical Pricing and Reimbursement Information); 2011. Available from: http://whocc.goeg.at/Downloads/Conference2011/PraesentationenPPRIKonferenz/Day2_morning_Sitzungssaal_1130_Baumgärtel.pdf
24. Baumgärtel C, Godman B, Malmstrom R. What lessons can be learned from the launch of generic clopidogrel? Generics and Biosimilars Initiative Journal (GaBI Journal). 2012;1(2):58-68. doi:10.5639/gabij.2012.0102.016
25. Abuelkhair M, Godman B, Fahmy S, Abdu S, Gustafsson LL. Challenges when introducing policies to engineer low prices for generics: experiences from Abu Dhabi. PPRI Conference Abstract Book; Vienna: PPRI (Pharmaceutical Pricing and Reimbursement Information); 2011. Available from: http://whocc.goeg.at/Downloads/Conference2011/Abstract_poster_book.pdf
26. WHO Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies. PPRI Conference. Final conclusions. Vienna: 2011. Available from: http://whocc.goeg.at/Downloads/Conference2011/PPRI_Conference_Conclusions_final.pdf
|
Author for correspondence: Sabine Vogler, PhD, Head of WHO Collaborating Centre for Pharmaceutical Pricing and Reimbursement Policies, Health Economics Department, Gesundheit Österreich GmbH/Geschäftsbereich ÖBIG – Austrian Health Institute, 6 Stubenring, AT-1010 Vienna, Austria |
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Related article
Generics policies–a globally-relevant implementation challenge

 [
[ [
[ [
[ [
[