Submitted: 4 November 2023; Revised: 24 November 2023; Accepted: 27 November 2023; Published online first: 28 November 2023
A study by Meijboom et al. aimed to compare the risk of and reasons for infliximab discontinuation between retransitioned patients and those remaining on biosimilar [1].
When the market exclusivity of the originator infliximab (Remicade) expired in 2014, many patients with inflammatory bowel disease (IBD) transitioned from the originator infliximab to an infliximab biosimilar, driven by cost containment reasons. Transitioning has been proven safe and effective in several double-blind studies, with numerous patients successfully making the switch in clinical practice.
However, approximately 7% of these patients subsequently retransitioned to the originator infliximab (i.e. they stopped the biosimilar and reinitiated the originator), mainly due to a (perceived) increase in disease activity or adverse events after transitioning to the biosimilar. It is unclear whether this sign of potential unsatisfactory treatment response is specifically related to the infliximab biosimilar, the patient, and/or the disease, including patients’ beliefs about the biosimilar.
Therefore, Meijboom et al. conducted a study with the aim of comparing the risk and reasons for infliximab discontinuation between two groups of patients: those who retransitioned to the originator and those who remained on biosimilar. The study included patients with IBD who had initially transitioned from infliximab originator to the corresponding biosimilar.
The risk and reason for retransitioning were assessed in all IBD patients who transitioned from infliximab originator to biosimilar between January 2015 and September 2019 in two Dutch hospitals. Retransitioned patients were matched with patients remaining on biosimilar (biosimilar remainder patients). The authors categorized patients’ reasons for discontinuing as either an unwanted response (i.e. loss of effect and/or adverse events) or as disease remission. A comparison of the risk of discontinuation due to an unwanted response was conducted between the two cohorts using a Cox proportional hazards model.
The study findings revealed that 22.7% of patients in the retransitioning cohort vs 13.4% in the biosimilar remainder cohort discontinued infliximab due to an unwanted response, and 2.3% vs 9.4% of patients, respectively, discontinued because their disease was in remission. The authors noted that retransitioned patients had more than threefold increased risk of discontinuing due to an unwanted response compared with biosimilar remainder patients (adjusted HR 3.7, 95% CI: 1.0–13.9).
Meijboom et al. further zoomed in on the retransitioned patients. About one in six retransitioned patients retransitioned due to objectively measured increased disease activity, e.g. elevated calprotectin and/or active disease seen on endoscopy, the other patients due to (subjective) symptoms only. Patients who retransitioned due to objectively increased disease activity discontinued their infliximab treatment more often than patients who retransitioned due to symptoms only (66.7% versus 23.7%).
In conclusion, the study demonstrated that patients who retransitioned were more likely to discontinue infliximab treatment due to an unwanted response compared to patients who remained on biosimilar. On the other hand, patients who remained on the biosimilar were more likely to discontinue infliximab due to remission. Patients who retransitioned had more than a threefold increased risk of discontinuing infliximab due to an unwanted response compared to patients who remained on the biosimilar. These findings indicate that retransitioning is mainly related to the patient and/or their disease, including the patients’ beliefs about the biosimilar. It is less likely to be related to the infliximab biosimilar itself.
The reason for retransitioning, which might have an impact on the course of infliximab originator treatment, is objectively measured disease activity or symptoms only, and it could be of importance in clinical decision-making. Clinicians could consider patients who opt for retransitioning to another treatment option.
Competing interests: The authors of the research paper [1] declared that there were no conflicts of interest.
Provenance and peer review: Commissioned; internally peer reviewed.
Reference 1. Meijboom RW, Gardarsdottir H, Becker ML, Movig KLL, Kuijvenhoven J, Egberts TCG, et al. Discontinuation of infliximab treatment in patients with inflammatory bowel disease who retransitioned to originator and those who remained on biosimilar. Therap Adv Gastroenterol. 2023 Sep 11;16:17562848231197923. doi: 10.1177/17562848231197923.
Author:Rosanne Meijboom, Trainee Researcher, Pharmacy Foundation of Haarlem Hospitals (SAHZ), Boerhaavelaan 24, 2035 RC, Haarlem, The Netherlands
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Submitted: 22 November 2023; Revised: 24 November 2023; Accepted: 27 November 2023; Published online first: 28 November 2023
Biologics are the fastest-growing class of medications in the United States and account for a substantial and growing portion of healthcare costs. The Biologics Price Competition Act of 2009 created an abbreviated approval pathway for the U.S. Food and Drug Administration (FDA) to help provide patients with greater access to safe and effective biological products. As of 1 November 2023, FDA has approved 44 biosimilar products, 7 of which are interchangeable biosimilars. These products can be used to treat many conditions such as chronic skin and bowel diseases, arthritis, kidney conditions, diabetes, multiple sclerosis, macular degeneration, and cancer.
Despite the rigorous requirements for comparative structural and functional analytical characterization data and one or more clinical studies that demonstrate a proposed biosimilar is highly similar to and has no clinically meaningful differences from the reference product, concerns of immune system mediated safety events associated with switching between biosimilars and their reference products persist.
Switching between biosimilars and reference products has been addressed in FDA guidance [1, 2]. As part of the demonstration of biosimilarity, a clinical immunogenicity assessment is expected to evaluate potential differences in immune responses and in some instances whether a single cross-over from the reference product to the proposed biosimilar would result in a major risk in terms of hypersensitivity, immunogenicity, or other reactions [1]. For interchangeable biosimilars, FDA guidance states that applications generally will include data from a switching study or studies and FDA anticipates that the data will be useful in assessing the risk, in terms of safety and diminished efficacy, of alternating or switching between the products [2].
Since the publication of these FDA guidances, experience with biosimilars and interchangeable biosimilars has increased considerably. While switching of biosimilars has been addressed in descriptive reviews, statistical methods have not been employed in a definitive fashion. This systematic review and meta-analysis [3] includes all of the identified randomized studies with one or more switches of biosimilars that were approved by FDA. Randomized controlled studies and their extension studies containing a switch treatment period (STP) to or from a biosimilar and its corresponding reference biological were identified from publicly available information maintained by FDA. These findings were augmented with data from peer-reviewed publications containing information not captured in FDA reviews. Forty-four STPs were identified from 31 unique studies for 21 different biosimilars. Data were extracted and synthesized according to PRISMA guidelines.
Meta-analyses were conducted to estimate the overall risk difference across studies. A total of 5,252 patients who were switched to or from a biosimilar and its reference biological were identified and 5,770 patients who served as no switch controls. Safety data, including deaths, serious adverse events, and treatment discontinuation showed an overall risk difference (95% CI) of -0.00 (-0.00, 0.00), 0.00 (-0.01, 0.01), -0.00 (-0.01, 0.00), respectively, across STPs. Immunogenicity data showed a similar incidence of anti-drug antibodies and neutralizing antibodies in patients within a STP who were switched to or from a biosimilar to its reference biological and patients who were not switched. Immune related adverse events such as anaphylaxis, hypersensitivity reactions, and injections site reactions were similar in switched and non-switched patients.
Safety and immunological concerns with switching between a biosimilar and its reference product, once or multiple times, have not been demonstrated in controlled clinical studies for FDA-approved biosimilars. This work adds to the growing body of evidence that switching between biosimilars and their corresponding reference products has not been associated with a greater risk of immunogenicity or safety concerns and is expected to reassure patients and their care providers. Regulatory recommendations on the need for studies with switches as part of the demonstration of biosimilarity and interchangeability are under review.
Competing interests: The authors of the research paper [3] declared that there were no conflicts of interest.
Provenance and peer review: Commissioned; internally peer reviewed.
References
Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product (May 2019) found at: https://www.fda.gov/media/82647/download
Guidance for Industry: Considerations in Demonstrating Interchangeability with a Reference Product (May 2019) found at: https://www.fda.gov/media/124907/download
Herndon TM, Ausin C, Brahme NN, Schrieber SJ, Luo M, Andrada FC, Kim C, Sun W, Zhou L, Grosser S, Yim S, Ricci MS. Safety outcomes when switching between biosimilars and reference biologics: A systematic review and meta-analysis. PLoS One. 2023;18(10):e0292231.
Author:Dr Sarah Schrieber, PharmD, Office of Therapeutic Biologics and Biosimilars, Center for Drug Evaluation and Research, U.S. Food and Drug Administration
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
The articles in this issue of the GaBI Journal highlight two important issues that must be considered when reading scientific publications. The first two articles illustrate the rapidly expanding influence of non-US, non-European economies on the global pharmaceutical industry. The second two articles illustrate the need to carefully evaluate the role of any potential author bias on an article’s content or conclusions.
The first Review Article by Alhomaidan et al. presents extensive observational data collected by the Saudi Food and Drug Authority (SFDA) of their pharmaceutical products approval and regulation procedures over the past 10 years. The authors describe the most prevalent product review gaps and good manufacturing practices (GMP) inspection deficiencies noted. They summarize the evolution of laws and standards that impact drug regulation, examine the use of new approval programmes and standards, and delineate the changes in the number of drugs approved from 2011 to 2021. The SFDA’s long-term objective is to, ‘establish itself as a global hub of regulatory excellence and hasten patient access to medications’. Saudi Arabia as well as some other countries in the Middle East and North Africa (MENA) region are already playing an increasingly important, outsized role in other aspects of the global economy. The influence of the MENA region on the global pharmaceutical industry is likely to only increase.
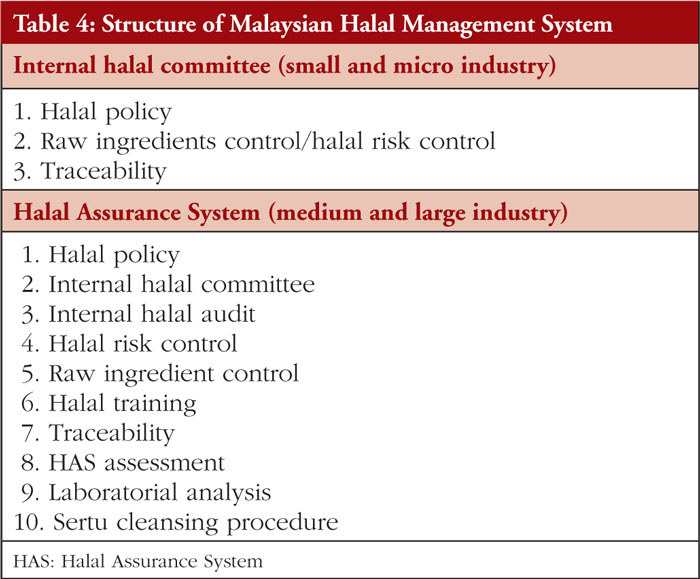
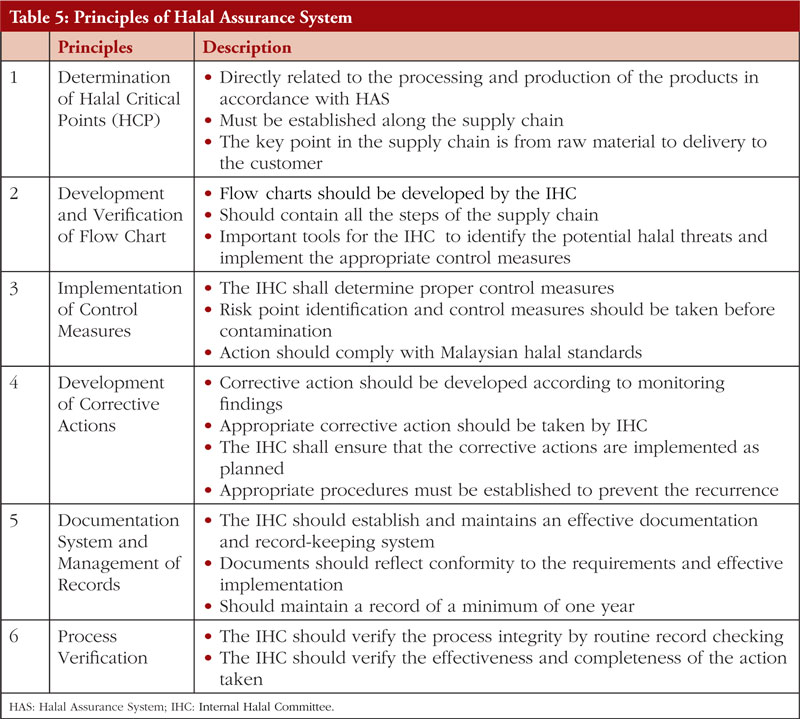
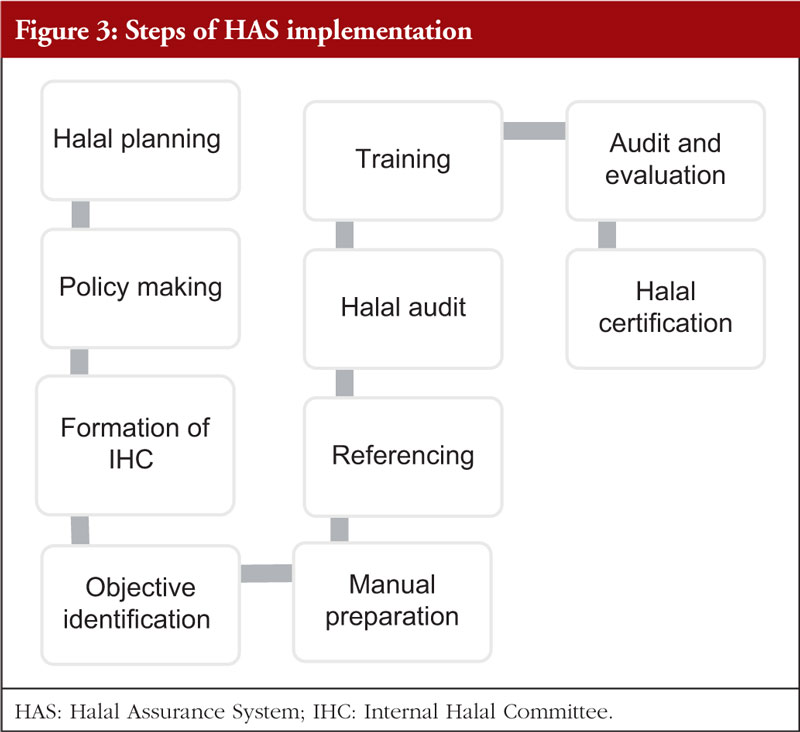
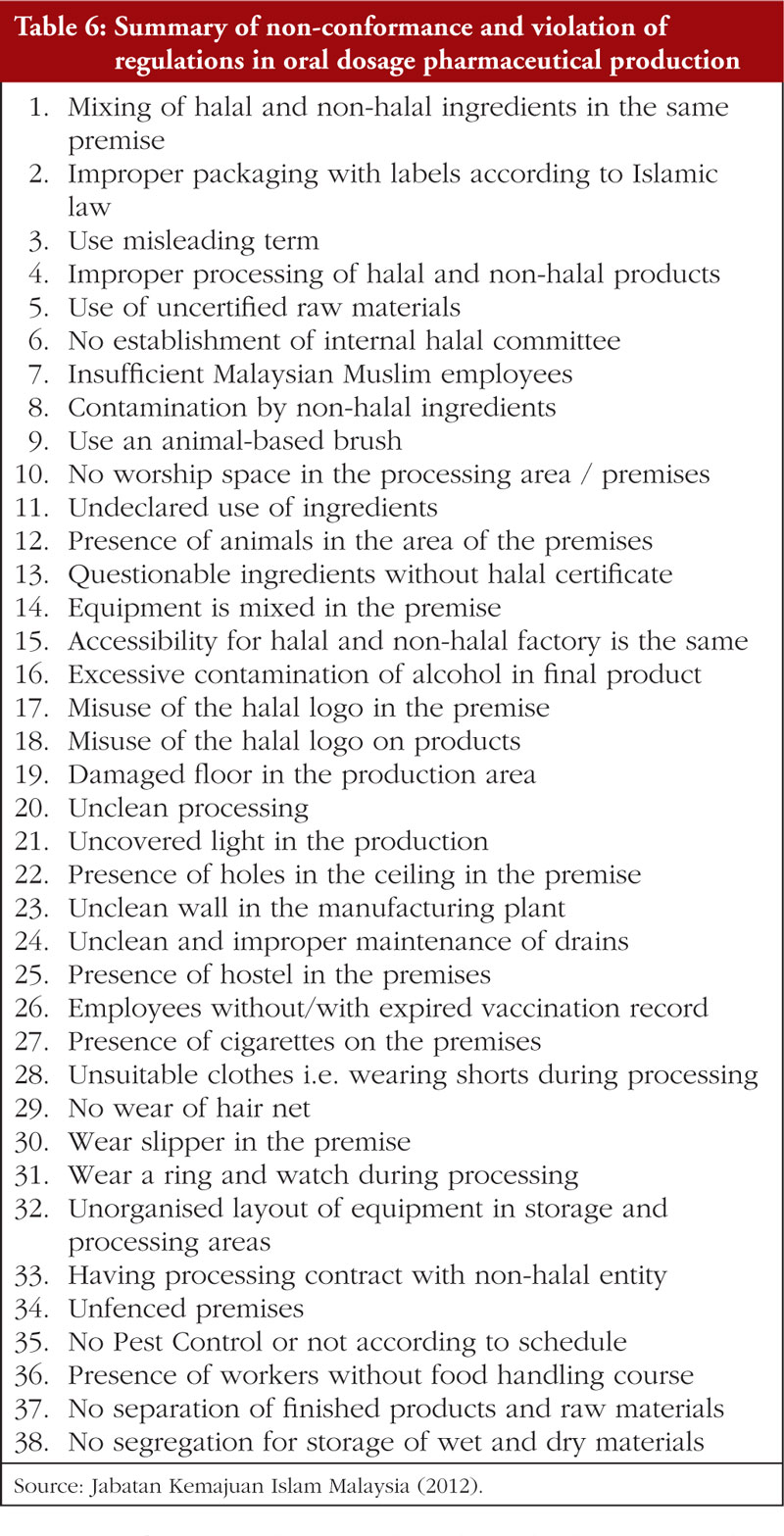
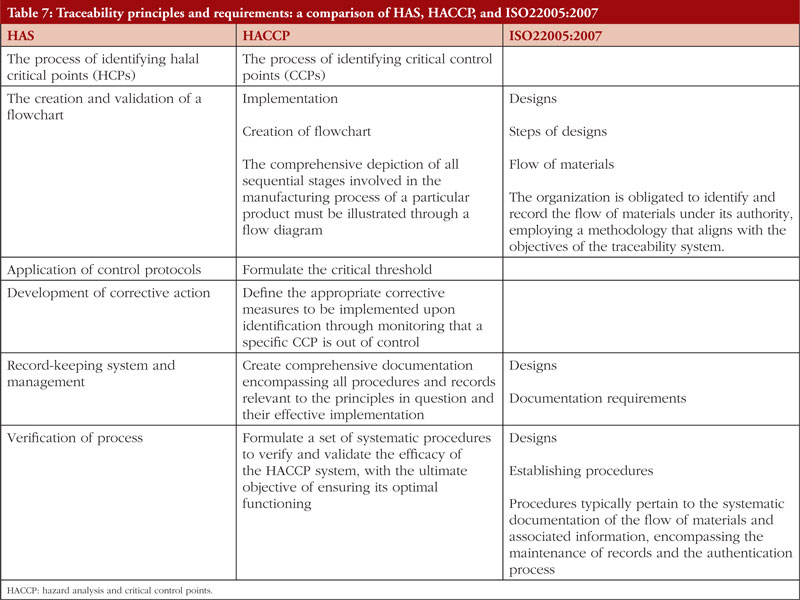
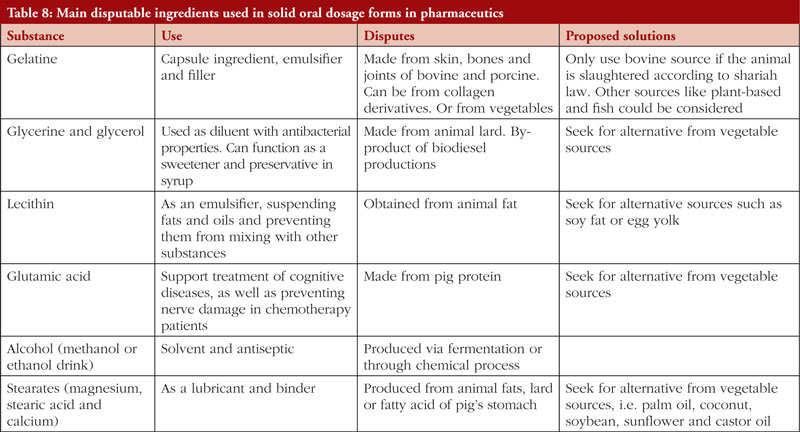
The second Review Article by Ka-Liong Tan et al. discusses the incorporation of a Halal Management System (HMS) by the rapidly growing halal pharmaceutical industry. The authors outline aspects of the HMS in the development and production of halal pharmaceuticals, explain the needs and requirements of an HMS, identify the challenges faced in implementation and establishing standardized certification, traceability, and effective recall mechanisms. The article should be of interest to any industry or company involved in the development, promotion, sale, or regulation of pharmaceutical products to the almost two billion Muslims in the world as well as to members of non-Muslim faiths that also follow basic halal dietary practices.
The Meeting Report by Reilly et al. presents in detail a summary of an online seminar sponsored by the Alliance for Safe Biologic Medicines (of which Mr Reilly is the Executive Director) and organized by GaBI staff. The meeting focused on the potential of the Inflation Reduction Act (IRA) to decrease innovation and new drug development as well as decrease patient access to some medications resulting from the price negotiations required by the act for a limited number of medications. Of note, the discussion focused solely on European and US effects of price controls. Much of the world’s population was simply not mentioned. The potential for the cost savings generated by the act to increase overall patient access to medications was also largely ignored. Finally, while no specific conflicts of interest were declared, there are numerous potential conflicts of interest raised by the relationships between the speakers and the pharmaceutical industry and other commercial interests that are likely to be financially adversely affected by the IRA. It is clearly important to consider all potential unintended consequences of legislation. I personally however wish that the meeting had included at least someone involved in the administration’s calculations of the potential positive economic and patient access implications of the act. It would also have been useful to present at least some data on non-pharmaceutical industry-based drug development methods such as those funded by governments and non-profit organizations, e.g. Gates Foundation, NIH, universities, WHO, governments.
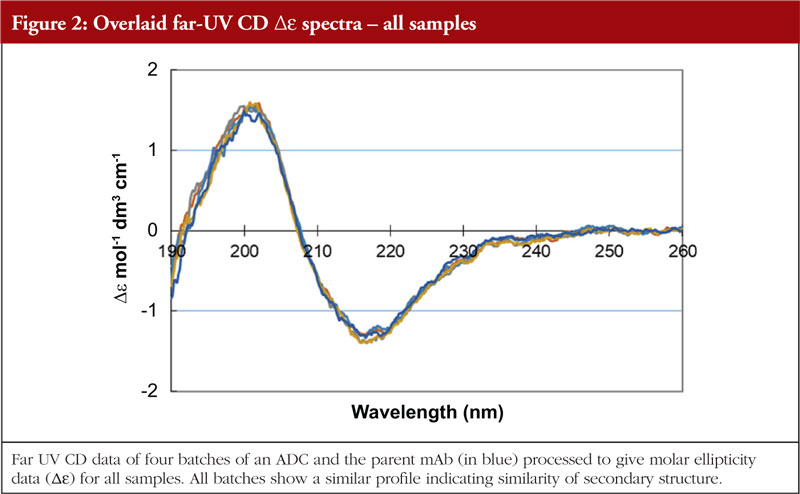
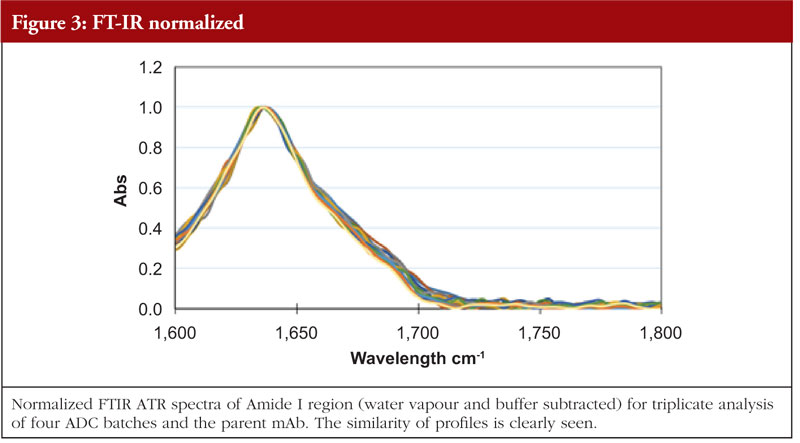
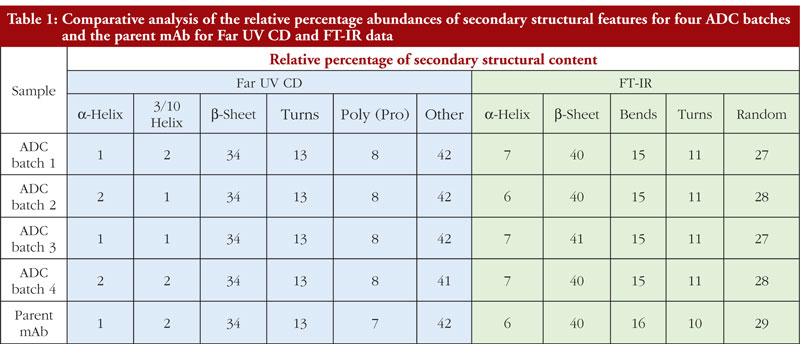
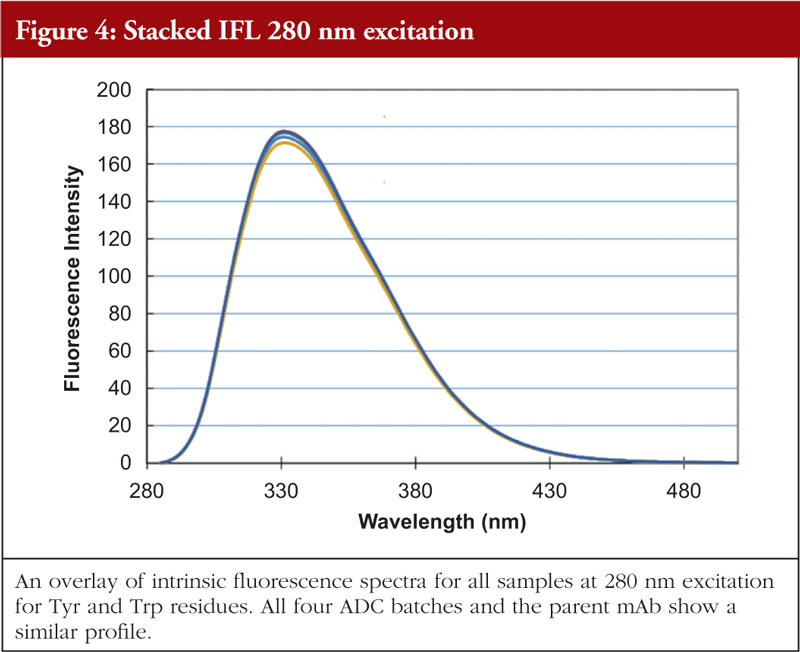
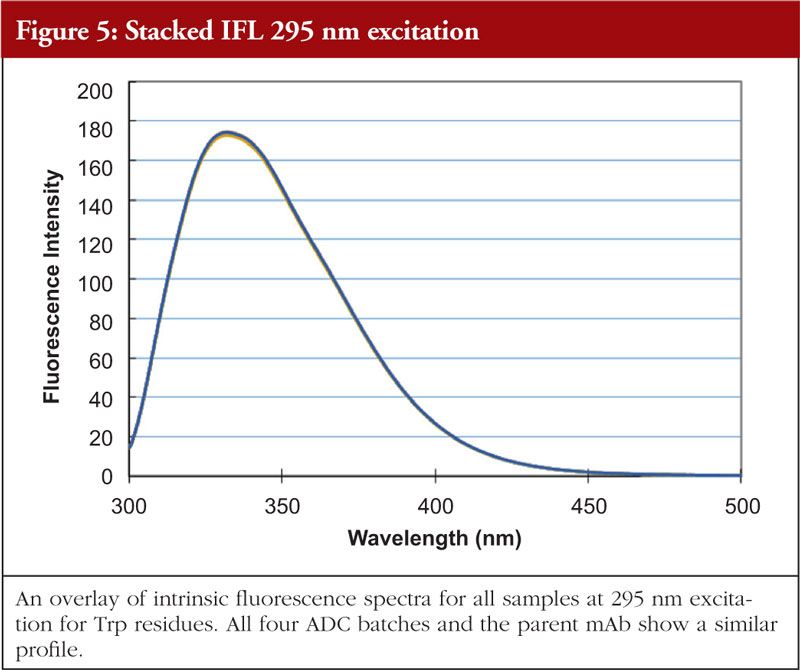
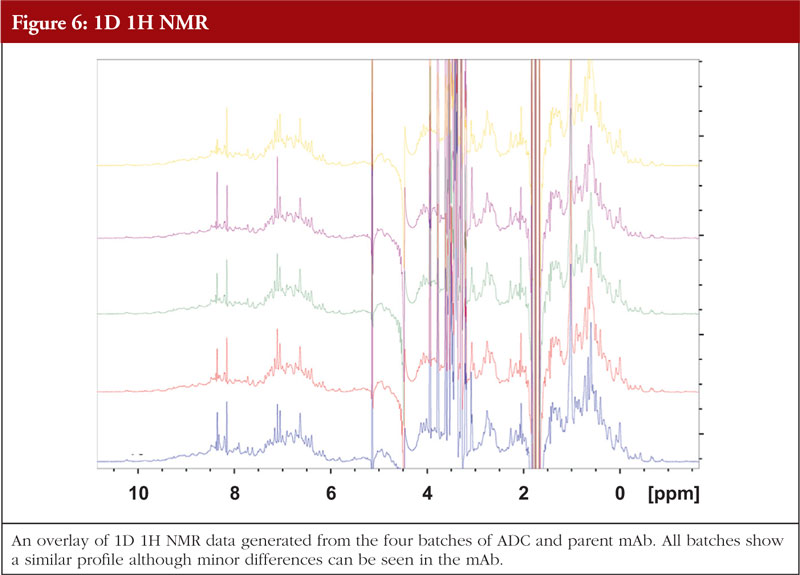
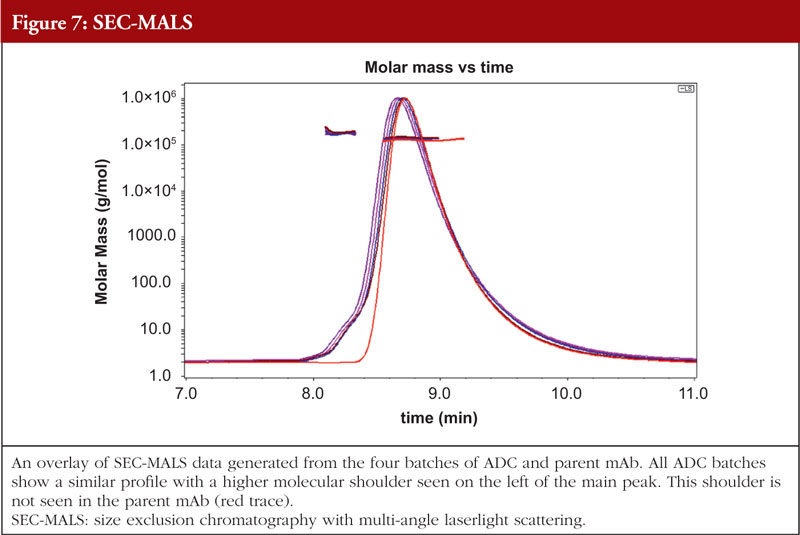
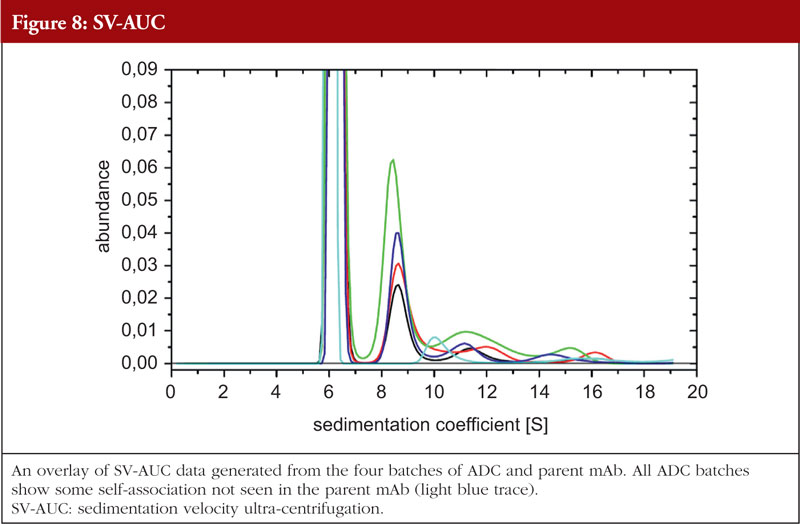
The final article in this issue by Dr Richard L Easton discusses various methods available to assess the higher order structure and aggregation of antibody drug conjugates, an interesting and promising class of biological methods. As an employee of a supplier of these methods to industry, the author’s potential conflicts of interest are clear. The presentation is; however, straightforward and (at least to me) unbiased. The acceptance of these methods by regulators will be critical in how they are incorporated into the regulatory process.
I realize it is difficult to be motivated to submit comments when faced with a world in which access to shelter, food and water is not guaranteed to an increasing number of the world’s population. Nevertheless, readers are encouraged to submit comments on these or any other topics.
Professor Philip D Walson, MD Editor-in-Chief, GaBIJournal
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Author byline as per print journal: Ali M Alhomaidan, PhD; Mohammed Abdulaziz Alageel, MSc; Turki Abdulaziz Alrafie, MSc; Hassan Mohammed Alqarni, MSc; Ibraheem Yahya Khbrani, MSc; Dalal J Alkhamis, MSc; Mohammed F Alkhalifah, MSc; Abdualmajeed S bin Jumaiah, MSc; Mohammed A Dahhas, PhD
Abstract:
The Saudi Food and Drug Authority’s (SFDA) pharmaceutical products approval and regulation procedures have changed and become more sophisticated over the course of the past ten years. Although medical advances give patients more therapeutic options, issues can arise if the promotion of pharmaceuticals prompts medications that were approved on the basis of limited evidence to be used in patients for whom there is little evidence of benefit or to replace tried-and-true therapies with better known risks and comparable or superior effectiveness. These flaws in the market can lead to costs that might not be justified by enhanced therapeutic effectiveness because new medicines are sometimes more expensive than the existing forms. As the top regulatory body in the Middle East and North Africa (MENA) region, the SFDA’s long-term objective is to establish itself as a global hub of regulatory excellence and hasten patient access to medications. In this paper, we discuss the most prevalent product review gaps (Common Technical Documents (CTD) deficiencies), good manufacturing practices (GMP) inspection deficiencies noted, explore the evolution of laws and standards that impact drug regulation, examine the use of new approval programmes and standards, delineate the changes in the number of drugs approved from 2011 to 2020, and expand the role and authority of the Saudi FDA.
Submitted: 25 September 2023; Revised: 17 October 2023; Accepted: 17 October 2023; Published online first: 31 October 2023
Introduction
All pharmaceutical goods that are commercially available in Saudi Arabia are required by law to have a marketing authorization [1]. The frequency of new medicine approvals has dramatically increased over the last decade. In Saudi Arabia, 5,498 pharmaceutical items were registered in 2011. By 2020, this number rose to 10,424. Drug approvals and the variety of active ingredients on which these new medications are based have been steadily rising over time as novel technologies are developed and introduced, regulatory frameworks change, and merger and acquisition activities fuel more consolidation. It is anticipated that these trends will persist over the coming ten years.
The use of unsafe, adulterated, or ineffective medications can lead to therapeutic failure, disease exacerbation, drug resistance, and occasionally even death [2]. This is why it is necessary to regulate the use of all pharmaceuticals. Additionally, the introduction of any unsafe or ineffective medication erodes trust in healthcare institutions, medical experts, and pharmaceutical producers and suppliers. Neither consumers nor governments should waste money on cheap, inefficient medications. To successfully oversee the production, distribution, and use of medicines in order to safeguard and advance public health, governments must set up powerful national regulatory agencies. SFDA, which was founded under Council of Ministers Resolution No. 1 from 07/01/1424 H, is responsible for assuring the correctness, quality, safety, and efficacy of medicines in Saudi Arabia, as well as the regulation and supervision of manufacturing facilities, importation, and registration of these products [3].
The task of conducting an open, prompt examination of pharmaceutical products for quality, safety, and efficacy poses challenges for pharmaceutical regulatory authorities. To establish a benchmark against which the impact of change can be measured and to develop realistic improvement initiatives, their performance in addressing that challenge should be routinely evaluated against established international qualitative and quantitative standards and recognized best practices and procedures. This will enable agencies to appraise their own performance and, ultimately, ensure patients have quick access to advanced, safe, and beneficial medicines.
GMP inspections are one of the tasks carried out by SFDA. This paper will explore some of the most prevalent flaws identified during inspections. The goal of sharing the inspection non-conformities is to give the industry the opportunity to evaluate the deficiencies found and subsequently address them as part of a self-continuous improvement effort. A company must request Saudi Food and Drug Authority (SFDA) to perform a GMP inspection visit (based on SFDA’s GMP guideline) when submitting a new product for registration and changing the location of production. Furthermore, a routine GMP inspection may be performed for manufacturers based on inspection priority for local manufacturers or to investigate complaints or recall requests.
Method
All medications that SFDA approved were identified and examined for this investigation. We reviewed all items registered between 2011 and 2021. The overall number, class, and most prevalent deficiencies of the drugs licensed each year were evaluated. Information on new molecular entities authorized by SFDA, biologicals, and generic drugs were all covered in this review. Additionally, the deficiency data pattern from the GMP inspections between 2018 and 2021 were examined. Hundred per cent of inspection deficiencies for inspections conducted between 2018 and 2021 were represented by the sample that was taken. This study primarily focused on inspection issues relating to production, material management, validation, premises and equipment, quality management, quality control, and personnel.
Results
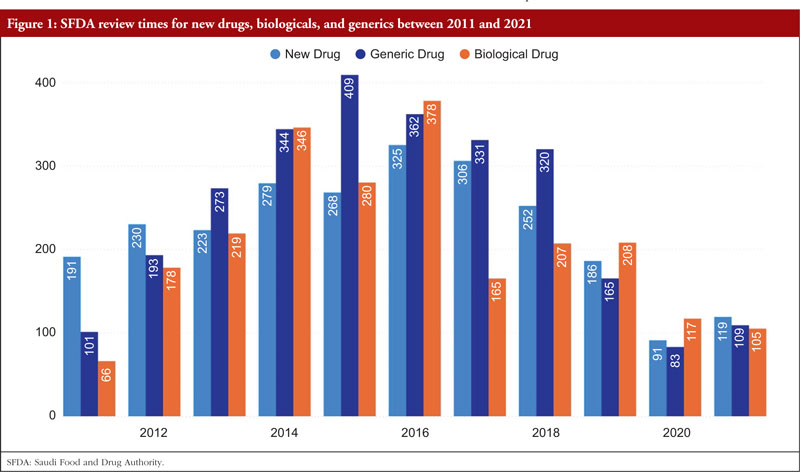
Reducing review timelines is a factor that helps pharmaceutical corporations achieve sales income more quickly while also allowing medications to reach patients more swiftly. The total number of days required by SFDA to examine new medications climbed from 191 days in 2011 to 325 days in 2016; however, it then rapidly decreased to 119 days in 2021. The total number of days required by SFDA to assess biologicals climbed from 66 in 2011 to 378 in 2016, and then fell to 105 in 2021. For all cycles of review, SFDA review times for generic pharmaceuticals grew from 101 days in 2011 to 409 days in 2015 and then decreased once again to 109 days by 2021,as shown Figure 1.
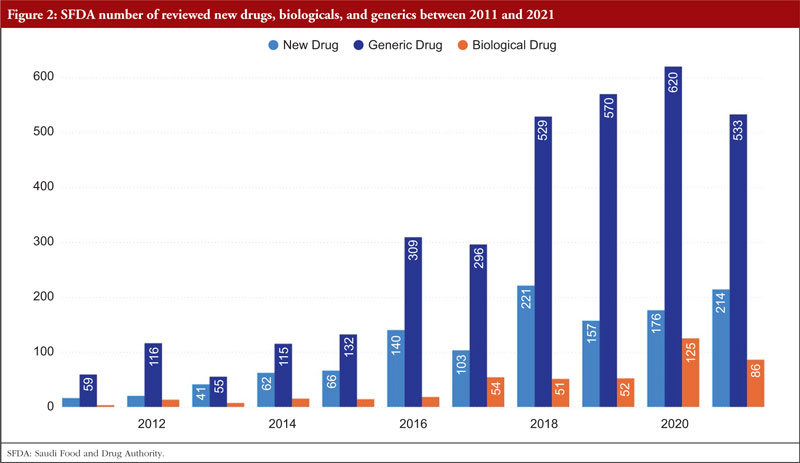
Over time, SFDA’s list of approved items has grown significantly. There were 214 new medications licensed in 2021 compared to 21 in 2011. With the number of generic pharmaceuticals approved annually rising from 59 in 2011 to 533 in 2021, the generic drugs pathway has been extensively employed. From 4 items in 2011 to 86 in 2021, the number of approved biologicals also grew, as shown Figure 2.
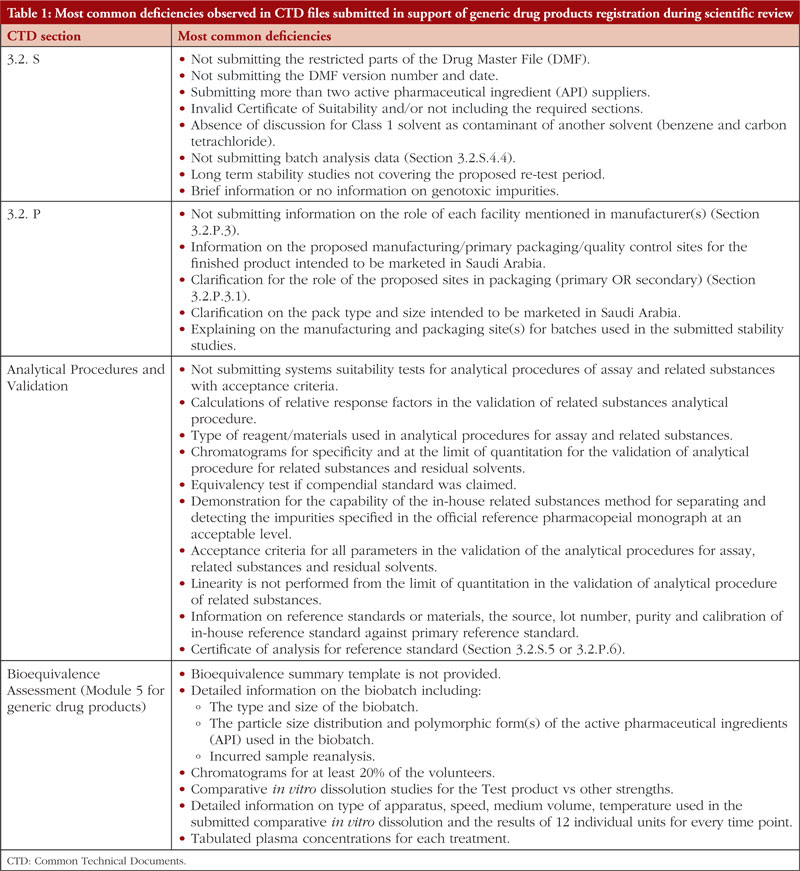
If SFDA did not approve a medicine during the first review cycle, it was mandated to send to the applicant an evaluation report outlining the shortcomings that the sponsor must correct. Here we list the most frequent deficiencies reported in the Common Technical Documents (CTD) files that were submitted in response to generic drug product registration during the scientific review of data as part of SFDA’s ongoing effort to streamline the review process and lower the number of deficiencies cited for the applications. These deficiencies are presented in Table 1. However, this list does not include every defect that has been found.
In total, 118 GMP inspections were conducted in 2018. Three producers of human-targeted pharmaceuticals and four veterinary medicine manufacturers were among the seven whose operations were suspended in 2018. Eighty-four of the 118 GMP inspections that were conducted in 2018 had major or critical defects. Twenty of the inspections with major/critical problems took place in Saudi Arabia, while 64 were performed abroad.
Twenty-three of the 124 GMP inspections conducted in 2019 were in Saudi Arabia, while 101were overseas. 2019 saw the suspension of eleven manufacturers. There were also 590 major deficiencies and 94 critical deficiencies identified in 2019.
2020 saw 43 GMP inspections, of which 12 were conducted abroad and 31 in Saudi Arabia. This reduction can be attributed to the COVID-19 pandemic. In total, 237 major flaws and 25 critical deficiencies were identified.
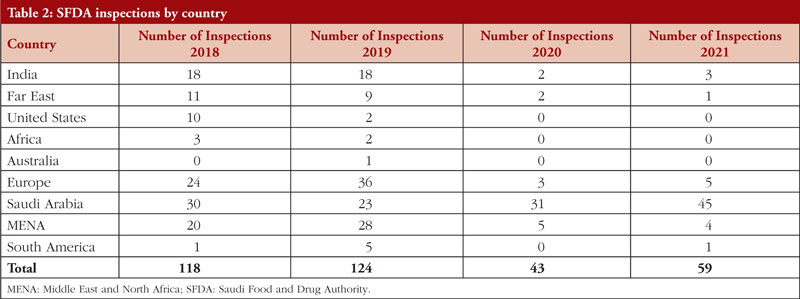
In 2021, 59 GMP inspections were conducted, with 14 being conducted abroad and 45 in Saudi Arabia (5 in Europe, 1 in China, 3 in India, 1 in South America, and 4 in the MENA region). There were 207 major deficiencies and 36 critical deficiencies. Between 2018 and 2021, SFDA performed 344 GMP inspections both within and outside of Saudi Arabia. The number of drug product inspections SFDA conducted between 2018 and 2021 is shown in Table 2 per nation.
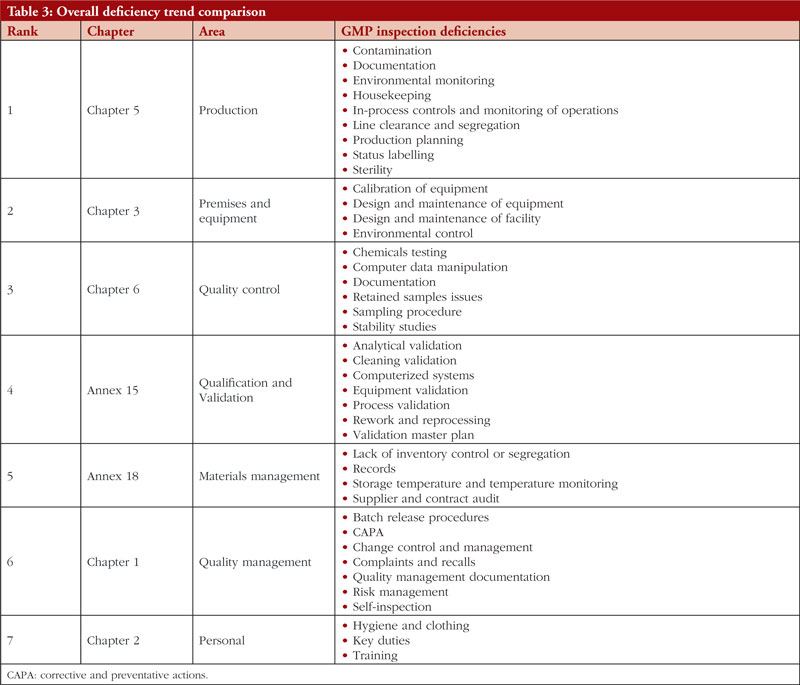
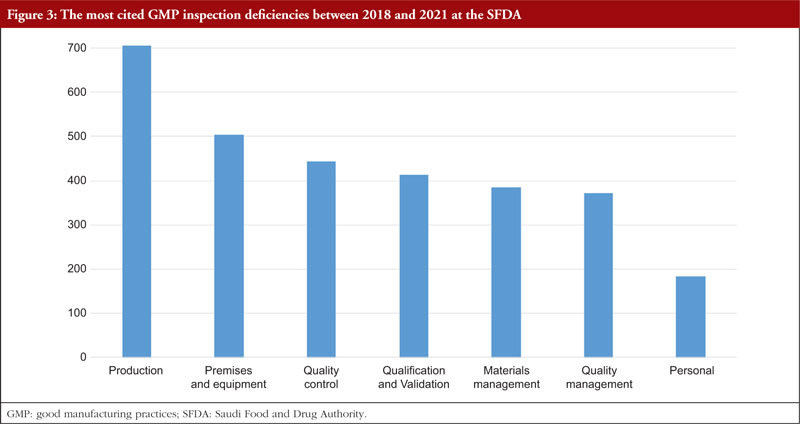
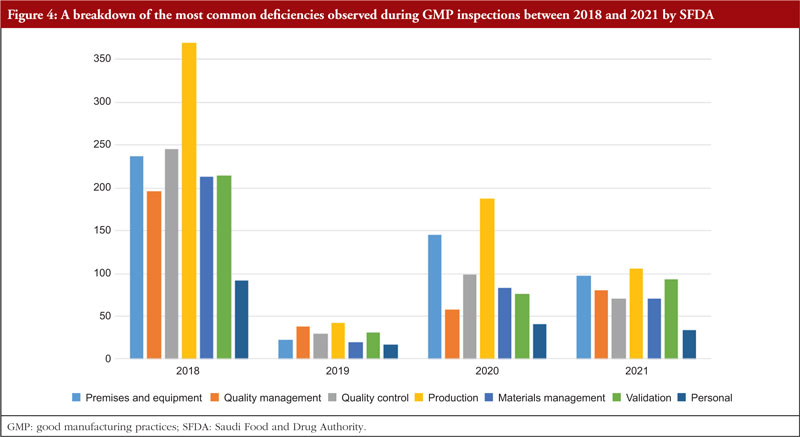
The trend is shown in Table 3, along with all classifications of deficiencies found in the top 7 chapters and annexes between 2018 and 2021. This data was derived from the GMP inspection reports. Between 2018 and 2021, the following, in order of frequency, are the most typical GMP flaws: Production, premises and equipment, quality control validation, materials management, quality management, and personnel. The most frequent flaws and a breakdown of them are shown in Figures 3 and 4.
Discussion
A crucial component of medication control is the registration of pharmaceuticals, commonly referred to as product licensing or marketing authorization. SFDA is required to license every drug that is sold, used, and distributed in Saudi Arabia. However, the quality and safety of products are assured by GMP inspection of the site, laboratory quality control testing and scientific examination of products prior to registration to ensure all marketed pharmaceutical goods fulfill the standards for safety, efficacy, and quality.
An effort is made to understand domestic and international trends related to drug evaluation as well as to obtain scientific knowledge. The most recent scientific discoveries serve as the foundation for evaluating pharmaceuticals. However, it is also important to take into account the context of the research studies that have been undertaken, their historical context, and previous choices regarding similar drugs. The decision on approval or rejection is based on a scientific and objective evaluation of the evidence supplied, taking into account the objective evaluation of benefits and hazards together with consideration of the patient’s perspective as well. Attempts are made to find the best solution for any issues that may arise during the new medication evaluation process by giving the applicant guidance and seeking understanding from many relevant parties after describing the cause and grounds for that specific concern. Efforts are invested in fostering mutual understanding with the applicant to support a smooth review, always keeping in mind the necessity to maintain open and honest communication at all times in order to secure a fair and unbiased stance.
SFDA approves over 900 medications annually. Many national regulatory bodies continue to confront difficulties as a result of resource limitations, despite their efforts to improve regulatory performance and work towards quicker clearance timeframes [2, 4]. The necessity for national regulatory agencies to use regulatory convergence initiatives, collaborative registration processes, and functional continental networks to carry out their regulatory mandates are brought on by growing workloads, developing technologies, and a lack of competence [5].
As can be observed in Table 1, this is by far the most active area in terms of common flaws and remarks made about applicants for generic drugs. The incidence of flaws illustrates how crucial the information regarding the controls suggested for the regular release and stability analysis of the drug product is. All suggested specifications (tests, procedures, and criteria) should be supported by solid scientific and regulatory reasoning, according to the applicants. This is not a complete list of the shortcomings in the sections on drug product release and stability, as was noted at the beginning of the study. However, the authors have made an effort to explain the fundamental causes of frequent deficiencies in stability testing and the control of therapeutic product. We want to explain why these flaws are mentioned and show how pharmaceutical development studies performed during the product’s original development could decrease the likelihood that these flaws become problematic.
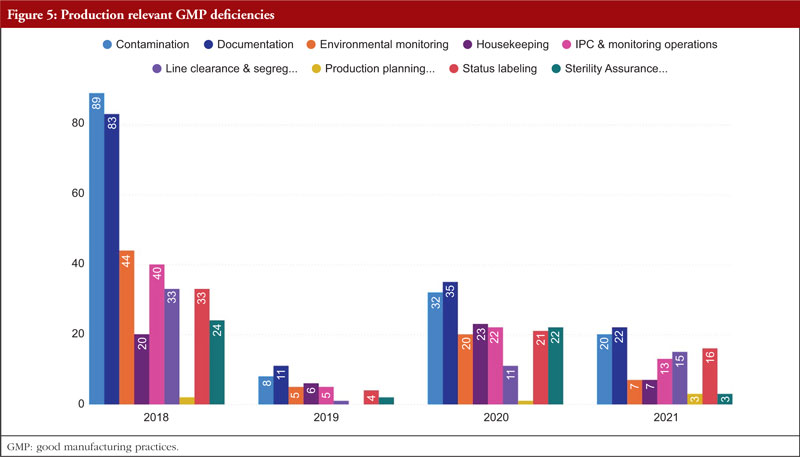
In regard to the faults found during the GMP inspection, we will begin by discussing Chapter 5, Production, as it is the source of the majority of the overall deficiencies, severe deficiencies, and major deficiencies. Chapter 5 is mentioned in almost 23% of all defects between 2018 and 2021. To produce high-quality products that satisfy regulatory criteria, production processes must adhere to GMP. Depending on the type of manufacturing undertaken, GMP for manufacturing operations includes criteria for the avoidance of cross-contamination during production, process validation, and environmental conditions. These GMP are required and should be supported by established and approved procedures and documentation for production activities [6]. Figure 5 shows production relevant GMP deficiencies.
During manufacture, sterile pharmaceuticals are more susceptible to contamination from particulate, pyrogenic, and microbiological sources. Special measures must be taken during the production of these products due to the health risk posed by using contaminated sterile supplies. All concerned personnel must possess the necessary knowledge, expertise, and training. Quality control is crucial, and production must adhere to rigorously established and approved preparation and sterilizing procedures. The environment in which aseptic procedures (such as equipment setup and filling) are carried out must be maintained in a controlled way and at an adequate quality in order to ensure product sterility. The handling of sterile materials before, during, and after the filling and closing, processes is an example of an activity that takes place in a crucial area [7].
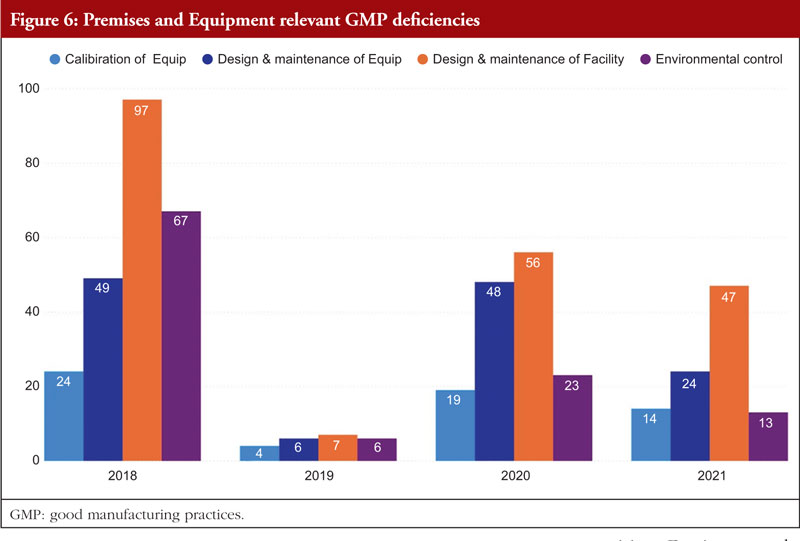
Chapter 3, Premises and Equipment, comes in second to Chapter 5 and is mentioned in almost 16% of all deficiencies observed between 2018 and 2021. The first requirement for ensuring that products are created safely is building design. Buildings must be of adequate size and designed in such a way to support these activities in order to allow cleaning, maintenance, and proper operations. In order to avoid cross-contamination between different sections, drug product containers, closures, labels, in-process materials, or drug products, manufacturers should have enough room for the orderly positioning of equipment and supplies. The unidirectional flow of parts, drug product containers, closures, labels, in-process materials, and drug products from minimally regulated regions to those with greater control should also be developed to reduce the risk of contamination. Each region of the production area that these materials pass through is under closer observation and supervision. For instance, the section where the bulk product is packed into containers will have more environmental controls than the loading dock at which raw materials and components are received [7]. Figure 6 shows Premises and Equipment relevant GMP deficiencies.
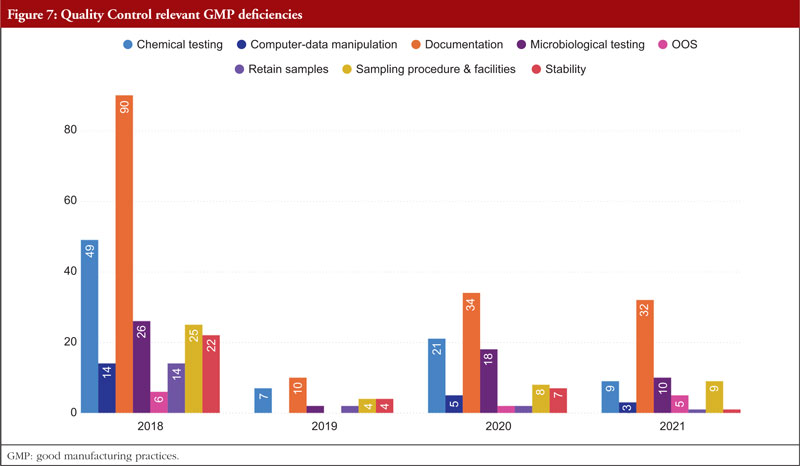
Third among the most often mentioned chapters and annexes between 2018 and 2021 is Chapter 6, Quality Control. The organization, documentation, and release procedures that guarantee that the required and appropriate tests are performed, and that materials or products are not released for sale or supply until their quality has been judged satisfactory are all concerned with quality control deficiencies. These deficiencies include sampling, specifications, testing, as well as organization, documentation, and release procedures. Quality control is not limited to laboratory procedures. It must be considered in all decisions that can have an impact on the product’s quality. It is thought that the successful operation of quality control depends on its independence from manufacturing. A quality control department should be present in every manufacturing site. The head of this department should have the necessary training, experience, and access to one or more control laboratories. It should be separate from other departments. To guarantee that all quality control procedures are successfully and consistently carried out, sufficient resources must be provided. Figure 7 shows Quality Control relevant GMP deficiencies.
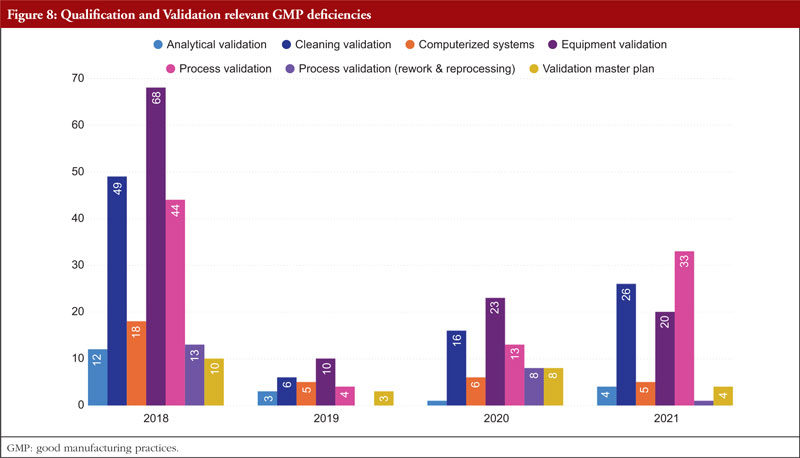
The next item on the list is Annex 15, Qualification and Validation. The validation of analytical results, cleaning validation, validation of computerized systems, validation of equipment, rework and reprocessing, and validation of processes are the most commonly referenced paragraphs. Any GMP facility, utility, piece of equipment, or process must be qualified and validated in the pharmaceutical industry. The goal of qualification and validation activities is to produce written proof that a facility’s utilities (such as water, gases, and air) and processes are created and run in accordance with SFDA’s GMP standards. Figure 8 shows Qualification and Validation relevant GMP deficiencies.
Appropriate certification and validation procedures must take place in order to guarantee that the facility, utilities, process, and equipment have been planned, constructed, deployed, and operated as intended. Design lays the groundwork for the overall success of a qualification and validation programme. For something to be built, installed, or manufactured, certain attributes must be present. These requirements are prescribed by specifications. The item being constructed, installed, or manufactured must meet specific requirements. You can categorize requirements as either user requirements or functional requirements. Specification and design requirements are focused on the characteristics that are essential to the quality of the final product and patient safety, and these criteria may be stated in the requirements and specification papers.
The manufacture and packaging of APIs and pharmaceutical goods must comply with regulations, which forbid both environmental and drug-related contamination. Equipment for washing and cleaning must be chosen and utilized with care to avoid becoming a source of contamination. The goal of the cleaning validation is to confirm the cleaning process’s efficacy in removing product residues, preservatives, degradation products, excipients, and/or cleaning agents, as well as in controlling any microbiological contamination. The manufacturer must also make sure that there is no possibility of cross-contamination of active components. To avoid impurities carrying over and building up, equipment needs to be cleaned at the proper intervals. The maintenance of cleanliness levels in the plant is partially ensured by a written cleaning process validation procedure [7].
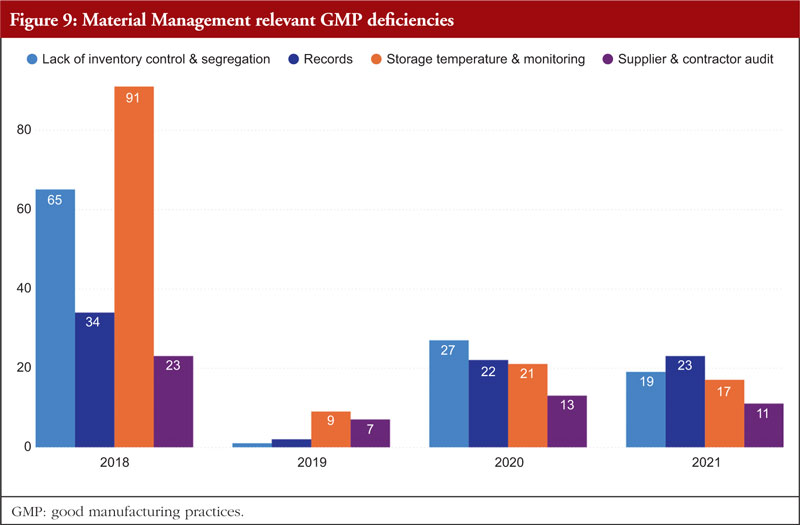
Material Management is next. The most frequently identified shortcomings include a lack of inventory control or segregation, problems with recordkeeping, problems with storage temperature and temperature monitoring, and audits of supplier contracts. Every document related to the shipping or delivery must be checked after receiving any material to verify accuracy and completeness. Excipients and active pharmaceutical components should only be sourced from recognized vendors for starting materials. Starting materials used in the production of APIs should be received in accordance with specific requirements in addition to those for starting materials used in medicinal products. It is best practice to confirm that incoming materials are correct, test them (if necessary), and then release them before combining them with current supplies. Figure 9 shows Material Management relevant GMP deficiencies.
Written sampling protocols that include the identities of the person(s) authorized to take samples, the techniques and tools to be used, the quantities to be taken, and any measures to be maintained to prevent contamination of the material or any degradation in its quality are preferable. All materials should have established sampling protocols and strategies that have been approved by the quality unit. Materials should be kept off the ground and placed in storage facilities with enough capacity to allow for orderly storage as well as to make cleaning and inspection easier. Any necessary label storage conditions, such as regulated temperature and humidity, should be maintained in storage locations. The monitoring equipment should be frequently examined and calibrated, and temperature monitoring records should be kept.
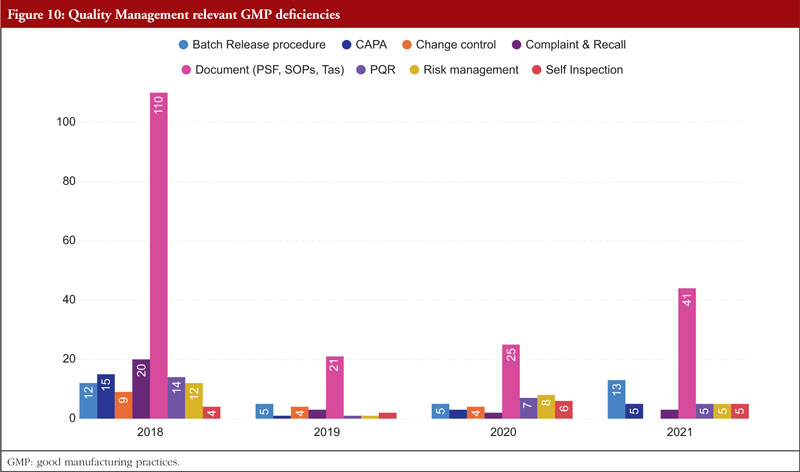
The next chapter is Chapter 1, Quality Management. The most frequently stated problems relate to batch release policies, CAPA, change control and management, complaints and recalls, quality management documentation, risk management, and self-inspection. Senior management is ultimately in charge of making sure a strong pharmaceutical quality management system is implemented that meets the quality goals. They are in charge of ensuring that the company’s duties, obligations, and powers are clearly stated, disseminated, and put into action. Figure 10 shows Quality Management relevant GMP deficiencies.
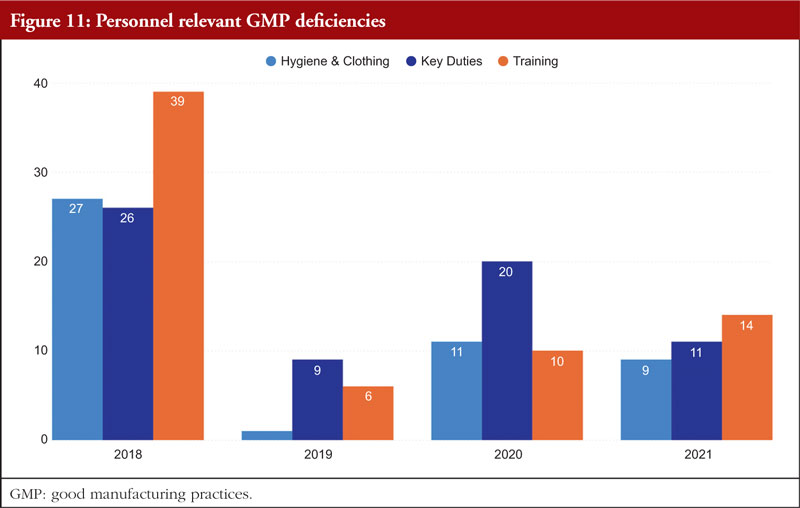
Chapter 2, Personnel, is the final chapter. Personnel must be properly educated, trained, and experienced in order to carry out their given responsibilities in accordance with GMP. A product may be declared adulterated by SFDA as a result of inadequate training. In such cases, the manufacturer would be prohibited from selling or distributing it. It is necessary to complete both GMP and specialized work task training. It is the responsibility of managers or supervisors to specify the training required for a certain position in a list of the necessary paperwork and educational programmes, such as a job curriculum. The curriculum can specify the scheduling requirements if frequent retraining is necessary; for example, as is the case with aseptic procedures. The training that must be completed before an employee can carry out a certain task should be specified in the curriculum, and the training completed should be formally recorded. Figure 11 shows Personnel relevant GMP deficiencies.
The provision of training should not be seen as a one-time event that occurs only when an employee is hired. When processes, batch records, and/or test methods are changed, or a task has not been completed recently, training pertaining to job tasks is required. Similar to this, when a worker’s position changes within an organization, his or her training, education, and experience should all be reviewed to ascertain what needs to be improved in order to accomplish the new duties. A current résumé or job history might be kept in the employee’s training file to show changes in work experience and education [7].
Conclusion
This paper examined the trends in SFDA medication approvals and GMP inspections from 2011 to 2020 and provided some insight into the most prevalent flaws in CTD dossiers that were examined for licensing purposes. The findings underline the significance of manufacturers leveraging such reports to pinpoint common sources of errors and strategize to prevent them in the future. By doing so, factories can enhance the quality and safety of their products while also maintaining compliance with regulatory standards. Furthermore, manufacturers can harness the insights gained from these generated reports to facilitate training for their employees. By analysing the documented errors and deficiencies, they can develop targeted training programmes to address these specific issues. This approach ensures that the workforce is equipped with the knowledge and skills necessary to avoid making similar mistakes in the future. By learning from past errors, manufacturers can create a culture of continuous improvement and maintain a proactive stance in preventing potential issues from arising. This integration of data-driven insights into training initiatives establishes a cycle of enhanced performance, minimized errors, and heightened product quality, ultimately contributing to the reputation and success of the company.
Disclaimer
The views expressed in this paper are those of the authors and do not necessarily reflect those of the SFDA or its stakeholders. Guaranteeing the accuracy and validity of the data is the sole responsibility of the research team.
Competing interests: The authors declare no competing interests for this work.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Ali M Alhomaidan, PhD Mohammed Abdulaziz Alageel, MSc Turki Abdulaziz Alrafie, MSc Hassan Mohammed Alqarni, MSc Ibraheem Yahya Khbrani, MSc Dalal J Alkhamis, MSc Mohammed F Alkhalifah, MSc Abdualmajeed S bin Jumaiah, MSc Mohammed A Dahhas, PhD
References 1. Saudi Food and Drug Authority. Royal Decree No. (M/108) dated 22/8/1441 AH Council of Ministers Resolution No. (534) dated 21/8/1441 AH. Law of Pharmaceutical Products, Pharmaceutical Establishments, and Herbal Preparations. 2020. [homepage on the Internet]. [cited 2023 Oct 17]. Available from: https://laws.boe.gov.sa/BoeLaws/Laws/LawDetails/3d191772-e60b-4925-b5e7-aba50097641f/1 2. Darrow JJ, Avorn J, Kesselheim AS. FDA approval and regulation of pharmaceuticals, 1983-2018. JAMA. 2020;323(2):164-76. 3. Saudi Food and Drug Authority. Royal Decree No. M/6 dated 25/1/1428 Cabinet Resolution No. 31 dated 1/24/1428, ‘The Law of The Saudi Food and Drug Authority’, 2007. 4. Tsiftsoglou AS, Ruiz S, Schneider CK. Development and regulation of biosimilars: current status and future challenges. BioDrugs. 2013;27(3):203-11. 5. Auclair JR. Regulatory convergence for biologics through capacity building and training. Trends Biotechnol. 2019;37(1):5-9. 6. Saudi Food and Drug Authority. Guide to good manufacturing practice for medicinal products. 2022 [homepage on the Internet]. [cited 2023 Oct 17]. Available from: www.sfda.gov.sa/sites/default/files/2022-08/SFDAGMPGuideline2022 7. The Saudi Food and Drug Authority. Guide to good manufacturing prac-tice for medicinal products. 2023. [homepage on the Internet]. [cited 2023 Oct 17]. Available from: https://www.sfda.gov.sa/sites/default/files/2023-01/SFDA-GMP-Guideline%20_0.pdf
Author for correspondence: Ali M Alhomaidan, PhD, Senior Consultant on Research and Innovation, Saudi Food and Drug Authority, 4904 Northern Ring Branch Road, Riyadh, Saudi Arabia
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Author byline as per print journal: Michael S Reilly, Esq; Thomas R Barker, Esq, JD; Charles M Clapton, Esq; Steven J Potts, PhD, MBA; Andrew Spiegel, Esq
Introduction: In the US, a Medicare drug price negotiation provision has been introduced in the form of the 2022 Inflation Reduction Act (IRA). An online webinar on the introduction of the IRA was held to discuss its implications on medicines innovation and impact on patient access to medicines. Methods: The webinar was held by the Alliance for Safe Biologic Medicines (ASBM) in collaboration with the Generics and Biosimilars Initiative (GaBI) on 26 July 2023. A number of expert speaker presentations were followed by a Q&A with the panel and the audience also had the opportunity to ask questions online throughout the webinar. Results: Presentations examined how the Medicare drug price negotiation provision in the IRA, can be considered a type of price-setting policy, similar to the European-style price control policies applied to medicines. In addition, the threat that government price setting poses regarding innovation and access to medicines for patients was discussed. Experiences with cost containment efforts in different countries and their impact on patient care and innovation were a key focus. Further details of the presentations were discussed during the Q&A, and clarifications made via the concurrent online Q&A. Conclusion: The webinar enabled an in-depth discussion of the Medicare drug price negotiation in the IRA and its potential negative impacts on innovation and patient access to new treatments. The US Medicare system’s current policy of private drug price negotiation versus European-style government price-setting were explored. Patient access to new drugs in the US, and resulting health outcomes, were compared favourably to that of European patients.
Submitted: 8 October 2023; Revised: 6 November 2023; Accepted: 8 November 2023; Published online first: 21 November 2023
Introduction
In 2022, the Inflation Reduction Act (IRA) (Public Law 117-169) was signed into law in the US. This is designed to empower the US Secretary of Health and Human Services (HHS) and enable them to develop and implement methods and a process to negotiate a limited number of maximum fair prices (MFPs) for prescription drugs in the Medicare programme, directly with manufacturers. At present, under the negotiation programme, the US government has selected 10 Part D drugs for negotiation for initial price applicability in year 2026 [1]; and this will scale up to 20 Part B and Part D drugs by 2028 [2]. The 10 Part D drugs are: apixaban (Eliquis), empagliflozin (Jardiance), rivaroxaban (Xarelto), sitagliptin (Januvia), dapagliflozin (Farxiga), sacubitril/valsartan (Entresto), etanercept (Enbrel), ibrutinib (Imbruvica), ustekinumab (Stelara), insulin aspart (Fiasp, Fiasp FlexTouch, Fiasp PenFill, NovoLog, NovoLog FlexPen, NovoLog PenFill). However, several drugmakers have sued the US government over the IRA [3, 4].
With this context, a webinar entitled ‘Drug price negotiations: impact on healthcare development and patient access to medicines’ was held by the Alliance for Safe Biologic Medicines (ASBM) in collaboration with the Generics and Biosimilars Initiative (GaBI) on 26 July 2023.
This webinar examined how the Medicare drug price negotiation provision can be considered a form of price-setting policy, similar to the European-style price control policies applied to medicines. It also sought to gain an understanding about the perceived threat that government price setting could pose regarding access to medicines for patients, from a healthcare providers perspective. Experiences with cost containment efforts in different countries and their impact on patient care and innovation were a key focus.
Methods
In this online event, held on 26 July 2023, the contributors discussed the creation of Medicare Part D, Medicare drug price provision and MFP determination. In addition, aspects of the US Medicare drug price negotiation system were compared to the system in Europe and discussions were held on whether the US government price setting diminished access to medicines for patients and the impact of the IRA’s small molecule penalty on cancer drug innovation.
The webinar was introduced by Michael S Reilly, Esq, Executive Director of ASBM, and moderated by Steven Stranne, JD, MD, partner at Foley Hoag LLP. Expert presentations were delivered by Thomas R Barker, Esq, JD, Partner, Foley Hoag LLP, USA; Charles M Clapton, Esq, Vice President, Federal Government Affairs, Gilead Sciences, USA; Matias Olsen, Public Affairs & Policy Manager, EUCOPE, Belgium; Steven J Potts, PhD, MBA, biotech drug developer, International Cancer Advocacy Network, USA; Professor Philip J Schneider, MS, FASHP, FASPEN, FFIP, Ohio State University; former Vice President of the International Pharmaceutical Federation (FIP); and Andrew Spiegel, Esq, Executive Director, Global Colon Cancer Association (GCCA), USA.
Results
Expert Speaker presentations
There were several expert speaker presentations, followed by a Q&A session and an in-depth panel discussion, moderated by Dr Steven Stranne.
Mr Reilly opened the webinar by noting that the introduction of the IRA brings several changes to Medicare Part D.
He outlined that Medicare part D is a federal programme providing outpatient prescription drug coverage for Medicare beneficiaries. It was implemented in 2006 as part of the Medicare Prescription Drug, Improvement, and Modernization Act of 2003. Enrolment in Part D is voluntary, with the exception of those who qualify for both Medicare and Medicaid, who are automatically enrolled to ensure they have prescription drug coverage. He noted that there is a 90% satisfaction rate amongst beneficiaries.
It was highlighted that price ‘Negotiation’ (Price Setting) for Part D was considered and rejected. Price Setting has long been known to have a negative impact on innovation, as it reduces the potential return on investment for drug manufacturers. The webinar set out to provide historical evidence of this effect in Europe and elsewhere. Ultimately, this could lead to fewer drugs for US patients in the future, as well as for patients worldwide, given that the US is the global leader in pharmaceutical manufacturing, responsible for producing more than 60% of the world’s new drugs.
The IRA builds on policy discussions dating back to the creation of Part D itself, granting ‘negotiation’ authority to the Centers for Medicare and Medicaid Services (CMS). The webinar provided a forum for experts from various fields to examine the potential effects of this policy on drug research and development, and patient access.
Mr Reilly emphasized that the IRA focusses on the price setting and ultimately, it does not look at value and outcomes and this is likely to lead this legislation to have a lot of negative effects and the impact of the IRA on patients and the patient community is key.
The ASBM has launched a microsite, IRApatientinfo.org, which aims to be a centre of information on IRA provisions and the aspects of IRA, as well as developments that impact the patient community.
Mr Reilly concluded that a second webinar will examine the effects of several other IRA provisions, including: 1) biosimilar entry (2-year pause); and 2) ASP+8% reimbursement.
Dr Stranne added that it is key to consider how to make sure the next generation (or generations) of patients have meaningful access to innovative technology that is life saving and life enhancing. He noted that the webinar panellists would provide a wide range of perspectives on how best to tackle the challenges in establishing fair and adequate pricing for innovative pharmaceuticals and biologicals. The webinar brought together perspectives of key stakeholders including the patient and family, pharmacy representatives and regulatory advocates who have spent their entire careers focusing on patient access. In addition, there were industry perspectives as well as perspectives from both the US and Europe.
The creation of Medicare Part D and why Medicare drug price negotiation is really a particularly flawed form of European price controls and what that means for patients
This presentation was given by Mr Thomas R Barker, Esq, JD, a former Commissioner of the Medicaid and CHIP Payment and Access Commission (MACPAC), an advisory body that provides policy advice to Congress and the States on the Medicaid and CHIP programmes. He played a key role in the implementation of every major health policy initiative enacted during his time at the HHS between 2001 and 2008, including the Medicare Prescription Drug Benefit (Medicare Part D) and the Medicare Advantage programmes. He also chaired policy briefings on Medicare and Medicaid policy at the HHS, the Office of Management and Budget, and the White House.
The creation of Medicare Part D Historical context Mr Barker initiated the discussion by providing an introduction to the historical background of Medicare, particularly focusing on the year 2003 when the Medicare Modernization Act (MMA) was enacted.
He highlighted how the MMA introduced an alternative strategy for regulating costs and managing expenditure within the Medicare program, particularly in Part D. Additionally, Mr Barker noted how the IRA reversed the approach that had been previously embraced in the MMA for regulating prescription drug prices.
Since the enactment of the Medicare and Medicaid programmes in 1965, Congress and the Executive Branch have struggled to control healthcare spending in the Medicare and Medicaid programmes. In 1983, Congress and the Reagan Administration agreed on a new payment system for hospitals inpatient perspective payment system (IPPS) that was designed to control costs. Congress and subsequent Administrations adopted similar prospective payment models between 1987 and 1997 for other classes of providers. However, these payment models failed to meaningfully control costs. For example, Medicare payments to hospitals were US$36 billion in 1980; in 2021, they had grown to US$450 billion, an increase of 1,150%. Core inflation during that four-decade period, by contrast, was 280%. For virtually every category of Medicare services (hospitals, physicians, home health, skilled nursing, dialysis), the government strictly regulates pricing. The government sets a rate of payment and providers must accept that payment rate as a condition of participating in the Medicare programme. Mr Barker noted that the IRA uses the misleading phrase price negotiation while there is no negotiation. The Medicare payment rate is a take-it-or-leave-it price. Providers have a right to comment on proposed payment policies, but at the end of the day, the government establishes a set price for each medical procedure. Thus, the term price negotiation is a misnomer, it is going to be like every other payment system in the Medicare programme where CMS establishes the rate of payment and providers, Part D plans and pharmaceutical and biological manufacturers will be forced to accept that price given the history of price controls in the Medicare programme. CMS has final authority to set the rate of payment and providers, health plans and pharmaceutical and biological manufacturers will be forced to accept that price. Unlike every other payment system in Medicare, however, MFP gives CMS an unprecedented (and poorly defined) mandate to set the price based on a subjective ‘lowest maximum fair price’ standard that is not applied in other healthcare sectors.
Part D: a different model Mr Barker then introduced the concept of Part D. When Congress created the Part D programme in 2003, it had the benefit of two decades of experience with healthcare provider rate setting. There was general consensus among both political parties, going back to the Clinton Administration, that if Medicare was going to adopt an outpatient prescription drug benefit, there needed to be a new model to control prices because government rate-setting had failed to control prices in all other sectors of the healthcare marketplace.
It was also highlighted that when President George W Bush signed Part D into law in December 2003, it relied on an entirely different model than the rest of Medicare:
The benefit would be run by private insurance plans
Negotiation for prices paid to pharmacies for drugs dispensed to Medicare beneficiaries would be conducted by pharmacy benefit managers and not the government
The government would be prohibited from instituting a formulary of covered drugs, from negotiating drug prices and from interfering in negotiations between Part D plans, manufacturers, and pharmacies
In summary, Part D has been enormously successful at controlling costs. When Part D was enacted in 2003, Congress estimated that spending on Part D drugs during the period of 2004–2013 would be US$770 billion. Actual expenditures were 45% lower, amounting to US$421 billion. The average monthly premium for part D in 2023 is US$32. In 2006, the average monthly Part D premium was also about US$32. By contrast, the Part B premium in 2006 was US$88.50; but it is now US$165 in 2023. This shows that the private marketplace is far more effective at controlling costs for the government and for beneficiaries. Notably, the Part B drug payment programme has successfully relied on a competitive market-based price to balance access and affordability, which has kept Part B drug inflation in check.
The IRA drug price ‘negotiation’ process will not be a negotiation at all Mr Barker noted that the so-called IRA negotiation process will simply substitute government rate setting with private market negotiations that have been enormously successful in limiting premiums and Part D expenditures. In that sense, the process is simply the European version of price controls. Government rate-setting of Medicare payments for other services has not been successful in controlling Medicare spending over the past four decades. Consequently, there is no reason to believe that a government-run rate-setting process will be at all successful in lowering programme costs. The Part D benefit redesign that was included in the IRA will provide benefits for enrollees, but those benefits have nothing to do with the drug price negotiation provisions of the law.
Why Medicare drug price negotiation is really European price controls and what that means for patients In his second presentation, Mr Barker discussed the effect of Medicare drug price negotiation on patients and other factors to increase the Part D spending.
1. Reductions in innovation There is going to be a reduction in the innovation of medicines. Manufacturers will be less likely to invest in new breakthrough therapies because the loss of profit incentives, which is built into the system, will go away under the IRA drug pricing policies.
More alarming, some members of Congress are proposing to apply the IRA’s government price-setting policies to launch prices of innovative products. This will further stifle innovation.
2. Effect on premiums Although it seems counter-intuitive, the price-setting provisions in the IRA may cause premiums to increase.
Part D premiums have been essentially flat over the past 20 years, while premiums in other parts of Medicare have escalated at rates equal to or even higher than inflation.
3. Other factors at play are likely to increase Part D spending Top-line statistics will show that Part D programme costs have increased more rapidly than in prior years by other factors. For example:
New GLP-1 agonists are going to drive programme spending. Here, the drugs are now being used for weight loss and physicians are prescribing them for haemoglobin A1c (HbA1c) and diabetes control, which will drive up Part D spending
New Alzheimer’s agents (especially if reformulated as Part D drugs) are going to drive programme spending
New gene and cell therapies (especially if reformulated as Part D drugs) are going to drive programme spending
In conclusion, Mr Barker stated that government price setting in the Medicare and Medicaid programmes have historically been ineffective in controlling programme costs. The free-market approach adopted on the enactment of Part D 20 years ago has clearly demonstrated the superiority of a private market approach to control Medicare spending. He warned that government rate setting of prescription drug prices will hinder innovation and ultimately harm patients.
Introduction of Medicare drug price provision and maximum fair price (MFP) determination
This presentation was given by Mr Charles M Clapton. Mr Clapton has nearly two decades of Capitol Hill experience, Notably, he served as health policy director for the Senate Committee on Health, Education, Labor, and Pensions, aiding the passage of the US Food and Drug Administration Safety and Innovation Act (2012). He also played a pivotal role as a lead Republican staffer during the Affordable Care Act’s congressional deliberations. He impacted the House Ways and Means and Energy and Commerce Committees by shaping Medicare Part D prescription drug benefits, revise drug payment methods for Medicare Part B, and Medicaid changes in the Deficit Reduction Act of 2005.
Medicare Part D The intent behind Part D was to create a market-based alternative to government price setting. Herein, the MMA established a programme where private plans negotiated with manufacturers and provided competing choices to patients, allowing beneficiaries to choose the plan that best met their needs. The Part D benefit was very successful, particularly in holding premiums essentially flat for almost two decades. At the same time, some beneficiaries have faced significant increases in out-of-pocket costs.
Ex-US price controls – limitations on patient access Prior to the enactment of MMA, and at an accelerating pace since then, many other countries particularly those in the Organisation for Economic Co-operation and Development (OECD) countries have implemented significant price controls on innovative drugs, or ex-US price controls. These price controls have achieved significant discounts, often well below the prices available in the US. These price controls have also resulted in patients in those countries facing limitations on access to innovative new medicines. This includes drugs not being approved as well as coverage not being provided for significant periods of time after the same drugs are available to US patients. This unfortunate outcome for patients is a foreseeable consequence of certain price control policies that have been implemented. It is worth noting that both Democratic and Republican lawmakers share a common frustration regarding these price controls. This has also led to a dynamic where the total costs of developing innovative new medicines have been shifted, so that the US healthcare system and patients are bearing an ever-greater share of total research and development (R & D) costs across the industry.
Inflation Reduction Act Anger over ex-US drug prices, increasing out-of-pocket (OOP) costs, coupled with support for expansion of Medicare price setting authority ultimately led to enactment of the IRA in 2022.
There are three main components of the IRA:
Inflation penalty – this imposes a cap on the level of price increases the manufacturers can take
Redesign Part D benefit (to address OOP issue)
‘Price Negotiation’ – a mechanism that would allow the government to negotiate prices
Generally, negotiation is a misnomer, it is really government price setting. Medicare will directly negotiate an MFP for a subset of brand drugs without generics or biosimilars competition. Negotiation will occur nine years after launch for small molecule drugs, 13 years for biologicals.
The bill’s creators picked these arbitrary dates of 9 and 13, and the reasoning behind their selection is not clear. However, this choice has potentially resulted in unintended consequences, which have tangible effects on the future of drug development. This brings us to the topic of negotiation mechanisms.
In 2026, the first year of price negotiation, 10 Part D drugs will be subject to negotiation. By 2030, this will have expanded to include 80 drugs subject to negotiation, covering both Part D, which includes small tablet drugs from a pharmacy, as well as Part B, which primarily consists of physician-administered injectables. Penalties for refusal to accept the ‘negotiated’ price are significant tax penalties for manufacturers, which, in fact, are excise taxes that can rise up to as much as 95% of total revenues of the drug. This essentially compels any manufacturer to participate in this negotiated price mechanism. This brings us back to why ‘negotiation’ is a misnomer, considering the concept of negotiation.
Negotiation process The negotiation process is a misnomer. What is going to happen is that manufacturers will be required to submit several specified types of data.
CMS will consider the following data and then make determinations as to what they think the price of the drug should be:
research and development costs and the recoupment of those costs
unit costs of production
distribution costs
prior federal financial support for novel therapeutic discovery and development, e.g. did National Institutes of Health (NIH) make any investments or were there other federal grants involved in the development of medicine?
approved and pending patent applications
Food and Drug Administration (FDA)-recognized exclusivities, and certain other applications and approvals
market data, and revenue and sales volume data
The negotiation process has minimal requirements for patient engagement, with no formal stipulations mandating the inclusion of patient voice. This stands in stark contrast to the patient engagement and process requirements seen in European Health Technology Assessment (HTA) programmes.
Negotiation is not required to go through rulemaking, it is conducted through sub-regulatory guidance and is exempt from judicial review. Therefore, even if it appears arbitrary or in violation of basic due process requirements, unless there is a constitutional claim, judicial review is precluded. This is in sharp contradiction to the implementation of the original Part D benefit, where interested parties could submit comments, and those comments had to be considered in the development of the new law.
Impact and consequences The Congressional Budget Office estimated the IRA will save US$237 billion over 10 years. There is significant uncertainty on the levels of discounts that negotiation will result in. Several reports have already highlighted the likely potential impact the IRA will have on future drug development. Vital Transformations, which is a research-based organization, worked very closely with trade organization BIO and PhRMA, recently issued a report that found that the IRA price controls will result in as many as 139 drugs over the next 10 years not being developed at all [5]. Academic researcher Tomas Phillipson at the University of Chicago, who served in a senior role during the prior administration in the White House, conducted research that showed similar negative impacts on development of new drugs [6]. The No Patient Left Behind Coalition has shared analyses highlighting the concerns of venture capital investors showing how the IRA is discouraging investments in certain types of drugs [7].
In conclusion, these analyses indicate that the IRA will disproportionately discourage future investments in and development of certain types of drugs:
Small molecule drugs – manufacturers will begin shifting their focus away from making future investments in small molecule drugs and instead, they will tend to invest in areas like biologicals, primarily due to the extended negotiation periods available
Subsequent indications for already approved drugs, for example, Keytruda, along with many in the field of cancer oncology, typically follow a model where they begin with a small patient group in a high level of unmet medical need. After gaining approval, they expand to earlier stages of treatment or larger indications and patient population. This approach is currently rewarded under the Hatch-Waxman Act, which offers extended exclusivity for new clinical trials and indications. However, under the IRA, there is a hard cap. For a small molecule drug treating glioblastoma, a nine-year countdown begins. Consequently, manufacturers will be disincentivized from conducting additional clinical trials to explore its potential efficacy against other cancer types
Rare diseases treatments.
Looking forward, the Biden Administration has already included a proposal in its FY 2024 Budget to accelerate price negotiation to apply to drugs at five years after launch and it would also increase the number of drugs subject to negotiation. Twenty-eight Senate Democrats have already co-sponsored the SMART Act which also accelerates negotiation to five years. In the longer run, federal budgetary pressures will insentivize Congress to expand price negotiation to apply at launch, using the mechanisms established by the IRA. This would further exacerbate the negative and unintended consequences on drug development.
Measuring the damage: impact of the IRA small molecule penalty on cancer drug development
Dr Steven J Potts, a successful biotech entrepreneur, discussed the impact of the IRA’s small molecule penalty on cancer drug innovation. Dr Potts has over 20 years of experience in the field of cancer, specializing in small molecules, complex cell therapies and biologicals. He contributed to the development of two approved drugs and supported molecular testing for entrectinib in 15 countries with 30,000 patients, a drug that obtained breakthrough designation equivalents in the USA, Europe and Japan.
Dr Potts’ presentation focused on the current effects of the IRA and how these effects (the level of damage) can be measured going forward. He highlighted that IRA imposes Medicare price controls on small molecules drugs at nine years after launch and on biologicals at 13 years, there seems to be no logical reasoning behind this difference. However, this distinction significantly impacts the ability of drug developer to secure funding from venture capitalists for small molecule development.
He also discussed how overall the IRA legislation reduces incentives for small molecules development compared with biologicals, because it only affects drugs purchased by Medicare [8-9], it also reduces incentives for drugs that treat seniors [10].
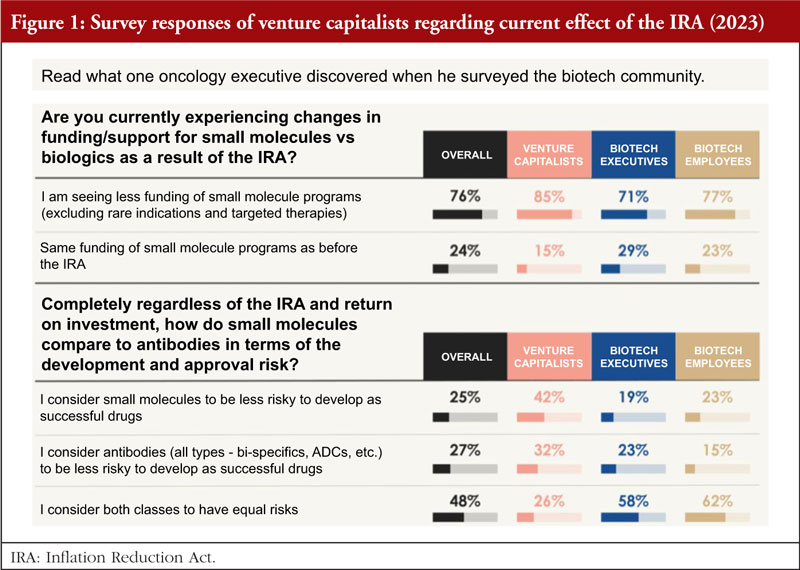
To better understand the current effects of the IRA, in March 2023, Dr Potts surveyed approximately 100 Venture funds in the pharma or biotech industry, asking two main questions, see Figure 1 [10]:
1. Are you currently experiencing changes in funding/support for small molecules versus biologics as a result of the IRA?
Eighty-five per cent said yes, they are seeing a significant decrease in interest in funding small molecules for large populations as a direct result of the IRA. This has a huge impact on fund raising for small start-up development in small molecules.
2. Regardless of the IRA and return on investment, how do small molecules compare to antibodies in terms of the development and approval risk?
Based on the survey results from March 2023, it is clear that both classes of drugs were considered to carry similar risks overall, 42% consider small molecules to be less risky to develop a successful drug, while 32% for antibodies; and 26% consider both classes of drug have equal risks.
The survey shows that six out of seven venture capitalists (VCs) have either withdrawn or been affected by the IRA, investment in small molecule drug development is shrinking and the vast majority of venture investors surveyed have moved away from funding small molecule programmes for Medicare patient populations as a result of the IRA.
In a similar survey on the impact of the IRA carried out by BioCentury with the support of BIO, see Table 1 [11], which was published around the same time as the Potts’ Survey of Venture Funds 2023 [10]. Key survey findings are:
The IRA is having a broad impact on the biopharma industry
Competitive landscapes will change, as companies are upending their pipeline and commercialization strategies
Expect pivotal changes in orphan drug strategies and modality choice
The IRA may accentuate the buyers’ market for partnering and mergers and acquisitions (M&A)
Sixty-six per cent of the respondents find the impact of the IRA ranged from existential crisis to major or minor changes, companies with revenues over US$1 billion see major or minor changes ahead; nearly one quarter (24%) are planning to prioritize their pipelines towards biologicals. All but one of these are US-based companies, 29% are weighing-up whether to shift to biologicals.
The IRA is particularly devastating in the neuroscience field, where timelines for development are already longer, success metrics lower and reimbursements not at all assured. The IRA will drive choice to invest in non-Medicare indications.
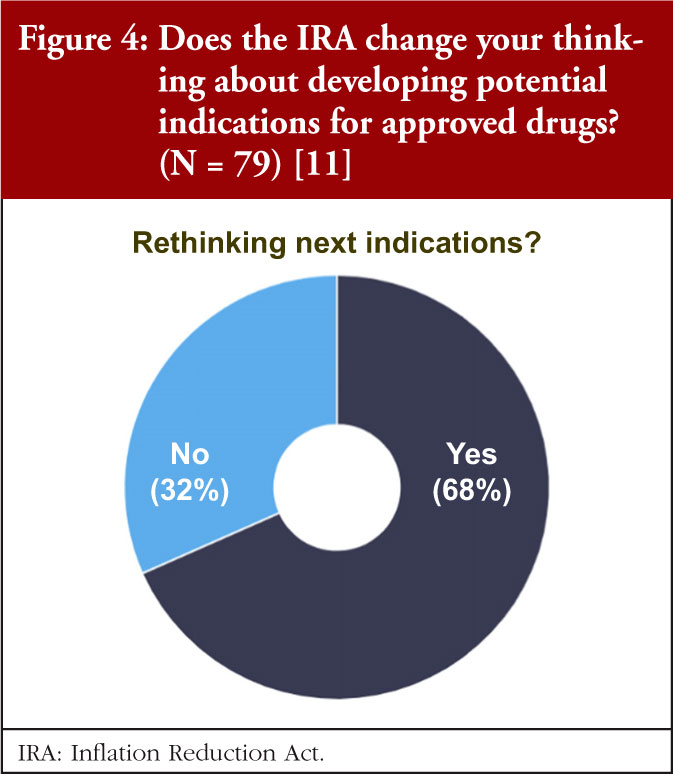
As one orphan indication per drug is the default, patients and companies may not get the most value possible from each drug. Because of provisions in the IRA, more than one third of the respondents will now prioritize orphan diseases over common, pursue one orphan indication instead of multiple, and develop distinct products for the indications, see Figure 2.
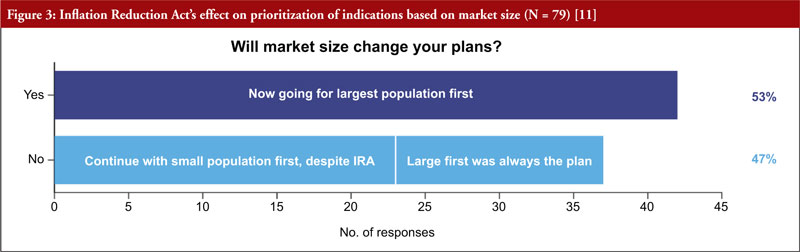
It was also clear that starting with small indications will fall out of favour, as more than half of respondents (53%) said they would go to market first in the largest population, see Figure 3. More than two thirds (68%) of respondents will rethink how to go about adding indications for approved drugs. Almost half (47%) of respondents are considering a new geographical strategy, considering to first commercialize their products in smaller indications abroad, and keeping large ones stateside.
Key points regarding the impact of the IRA on the pipeline strategy in the BioCentury 2023 survey:
Under the IRA, products will be subject to price setting nine years after the approval of new drug applications (NDAs) and 13 years after approval of biologics license applications (BLAs).
As a result, companies may be incentivized to seek approval in the broadest possible indication first to maximize total sales in the time before they become subject to price setting.
It may also discourage conducting trials to add new indications late in the window before price setting.
Orphan drugs are exempt from price setting but only if approved for a single orphan indication. This means approval of another indication can make the medicine subject to price negotiation.
The survey asked how these changes might affect respondents’ pipeline strategies, including their selection of indications and modalities.
Dr Potts asked, ‘Will ‘pipeline-in-a-product’ become a thing of the past?’, regarding this, the BioCentury data show that most are recalibrating the calculus for adding indications, see Figure 4.
More than two thirds of respondents say they will rethink how they go about adding indications for approved drugs. Parallel development will likely be the best way to target multiple indications in the allotted time window. For others, it may mean building a timeline of different indications from a single product is no longer commercially viable.
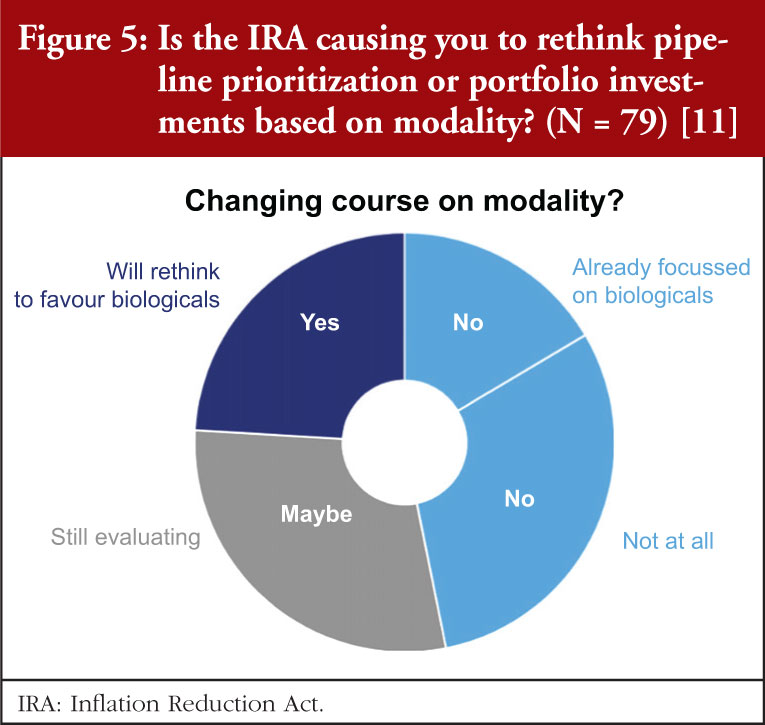
The survey data also indicate that we can expect to see a shift from small molecules to biologicals, see Figure 5. Nearly a quarter (24%) indicated that they were planning to prioritize their pipelines towards biologicals. All but one of these companies were US-based. Another 29% are weighing-up whether to shift to biologicals. A sizable minority (38%) have no plans to rethink their pipelines, with the balance (16%) already focused on those modalities.
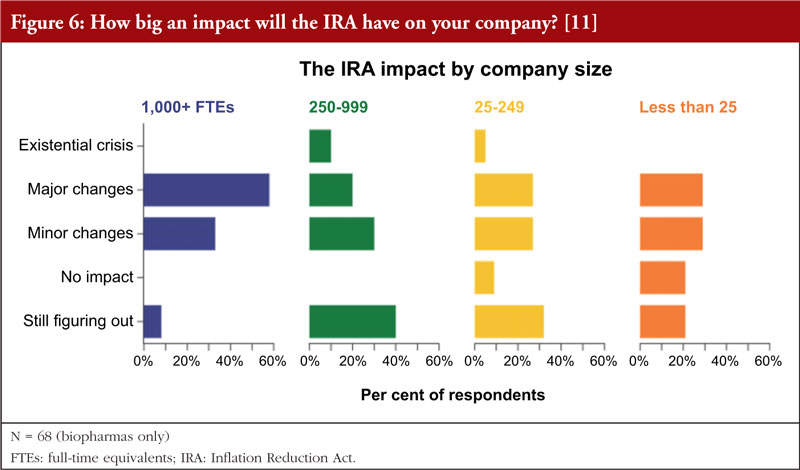
In addition, the data indicate that the smaller the company, the less relevant it sees the impact of the IRA, see Figure 6. The few companies that do not expect any impact were in the small to very small categories (<250 FTEs (full-time equivalents)). However, even among this group the majority (55%&ndash60%) expect major or minor changes. While 40% of mid-size companies (250&ndash999 FTEs) are still figuring things out the IRA impact, but the larger companies (58%) expect major changes.
In conclusion, these initial survey data and studies are ‘canaries in the coal mine’. Officials are requesting data on the impact of the IRA discouraging innovation in medicines, and they plead for further and more studies with data measuring the impact, challenges, and adverse effects on new drugs that may not be developed over the next six to 10 years because of the IRA shift unless it gets changed, particularly for small molecule drugs.
Cancer and neurological treatments, including Alzheimer’s disease with smaller indications, are going to be hit hardest by the IRA. It is possible that entire classes of innovation will be eliminated unless it is modified to grant both small molecule and biological classes 13 years prior to price setting. This is because it takes a long time for patients to change their medication, and no matter how good the drug is, sales always ramp up late. Therefore, those last four years will provide half of the profits.
Dr Potts provided resources of two revealing interviews with Dr Rafael Fonseca (Multiple Myeloma, Arizona) [12] and Dr Barbara McAneny (Solid tumour cancers, New Mexico, past President, American Medical Association (AMA) [13] on the impact of IRA on oncology patient care.
Government price setting diminished access to medicines for patients
Speaker Mr Andrew Spiegel, founder of the Global Colon Cancer Association (GCCA), an international patient advocacy organization representing colorectal cancer groups worldwide. He has over 20 years of experience in patient advocacy. Beyond colon cancer, Mr Spiegel advocates for healthcare policies globally. He chaired the fundraising committee for the International Alliance of Patients’ Organizations (IAPO) and co-founded the World Patients Alliance, the world’s largest patient organization. He is a founding member of ASBM.
In his presentation, Mr Spiegel highlighted that colorectal cancer is on the rise. The rate of colorectal cancer in Americans under 55 has nearly doubled since the 1990s. The rates among people aged under 40 are predicted to double by 2030. The American Cancer Society says there are likely to be around 153,000 colorectal cancer cases detected in 2023. It is already the second leading cause of all cancer deaths, behind only lung cancer, thus more and better treatments are needed urgently.
Unfortunately, the new drug pricing rules will harm cancer drug development. Under the IRA, Medicare will be able to ‘negotiate’ with drugmakers for lower prices on an expanding list of brand-name medications. Two types of drugs are affected, both are critical to cancer treatment, see Figure 7.
The IRA problem is that there is a ‘small molecule penalty’. As was previously pointed out by Dr Potts, the IRA makes small molecule drugs eligible for price controls nine years after FDA approval – four years sooner than biologicals. Those four additional years are critical for investors when deciding whether to invest in drug development. At present, it takes an average of more than US$1 billion and more than a decade to bring a new drug to market. Half of a new drug’s value is realized between its ninth and 13th years on the market, since less than 8% of drugs in clinical trials are approved, investors must ensure their rare successes pay off.
When it comes to the shift away from small molecule investment, this IRA policy will have serious consequences for cancer treatment. AstraZeneca, Merck and other biotech companies have already raised an alarm about how investment priorities will shift away from small molecule research in the coming years. Small molecule drugs happen to be the only way to target certain cancers as large molecule biologicals are too big to penetrate cell walls and attack cancers with intracellular targets. Mr Spiegel noted, ‘It’s not as if researchers always have a choice of which type of drug to focus on’.
In terms of the ‘negotiation’ aspects of the IRA, the process is to force manufacturers to ‘agree’ to a government dictated price, the IRA imposes an escalating ‘excise tax’ that begins at 186% of a medicine’s total sales revenue and reaches a maximum of 1,900%. As was brought into focus by previous speakers, this is essentially just price setting. During the process there is only one opportunity for patients and providers to weigh in, and it is only in writing, using a bureaucratic form with word limits that must be submitted shortly after selected medicines are announced. Key implementation decisions are also exempt from public ‘notice-and-comment’ procedures and from judicial review.
It is evident that government price setting hurts innovation. An example of a European case study is reduced drug development. In the 1970s, European companies developed most new drugs. However, since the implementation of price controls in Europe, 60% of new drugs are currently developed in the US, compared to 13% in Switzerland, 8% in the UK, and 6% in Germany and France [14].
In addition, price controls reduce patient access to cancer treatment. Of cancer medicines launched globally between 2011 and 2019, more than 96% are available to US patients. Only 65% are available in other developed nations such as Australia, Japan and the UK, which employ government price-setting policy. Mr Spiegel gave the example that Australian patients have lost access to drugs due to price controls in excess of what was agreed to by government [15]. This resulted in:
Fewer choices, not more choices for patients and physicians
De facto forced substitution as there are no available listed products other than the government-preferred product. Example: stage IV cancer patients now being forced switched
Concerns with long-term sustainability, i.e. reduced competition, product launches cancelled
‘Is America ready for European-style health outcomes?’, asked Mr Spiegel asked Mr Spiegel. Of new cancer medications, 90% are available to US patients within the first year of launch, whereas less than half of these are available to cancer patients in Canada, France, Germany and the UK [16]. Furthermore, cancer death rates per 100,000 are 1.6 to 1.8 times higher in Europe than those in the US [17]. If the US had European cancer death rates, it would mean more than 400,000 additional deaths each year from cancer. That would be equivalent to losing an entire city like Minneapolis, Oakland, New Orleans, or Tulsa – every year to new cancer death.
It is critical that patient communities stand up for innovation. For example, they can do this through education: Op-eds by patient advocates [18], outlines some good data on why the IRA is unfavourable for patients, and ASBM microsite: IRAPatientInfo.org – a centre of information on IRA provisions and the aspects of IRA, as well as developments that impact the patient community; and through legal challenges: Global Colon Cancer Association (GCCA) has joined a multi-stakeholder lawsuit with the National Infusion Center Association (NICA) and the Pharmaceutical Research and Manufacturers Association (PhRMA) representing the patient voice challenging the law [19].
Additional presentations were given by Matias Olsen, ‘Negotiating drug prices in the US-lessons from Europe’; and Professor Philip Schneider ‘Pharmacist perspective on the impact of drug price controls’.
Summary of panel discussions/Q&A
Following the presentations, the panel members discussed a number of questions. These discussions were moderated by Dr Stranne and are summarized here.
Question 1: How can the IRA be modified or fixed? In other words, what steps should be taken to improve existing policies to better represent the best interests of patients? In response, Mr Spiegel noted that it is hard to fix something once it has passed into law, and it would have been so much easier for the administration to listen to all stakeholders before the IRA was implemented. However, as it stands, what needs to be fixed is anything that stifles innovation, whether it is the number of years of protection or simply getting rid of the whole negotiation idea. The concept that one can actually negotiate with the US government must be discarded because, for one, it is not a negotiation, it lacks transparency and is misleading.
The lack of transparency in the IRA is a significant issue. There has been no way for patients and patient voices to become involved in the policy discussion, and there is no transparency about how these prices are going to be negotiated. The IRA is likely to result in measurable pullback from the industry, which will mean fewer drugs for patients. There needs to be an open way for patients to get involved and have a voice, but currently, it does not seem their viewpoints will be taken into account. This lack of transparency and the absence of patient/public input are why GCCA filed the lawsuit against the administration, along with other stakeholders.
Mr Reilly noted that the administration chose to go through the guidance process instead of the ‘notice-and-comment’ process with the changes to Medicare Part D and the IRA. In general, any major change in law within HHS would go through a normal 90-day ‘notice-and-comment’ process where it is necessary to seek input from the public and patient community, the industry and the provider community. This is necessary to try to work as partners to resolve issues that are likely to be contemplated by those who are being impacted by the law. However, when it came to the IRA, this was handled through guidance which is essentially 30 days and there is no requirement to listen to stakeholders. This demonstrates that the administration was not really open to getting input from stakeholders, going forward this is a major problem just in terms of how the IRA is perceived.
Mr Barker added that in 2004/2005, the US Department of HHS consulted extensively with the public during the implementation of the new prescription drug benefits. This included a notice and comment process involving patients, pharmacy benefit managers (PBMs), and manufacturers partnered to ensure the design of the benefit was correct. However, with the IRA, it seems the government does not want input from the public with their own fixed idea of how to do it and have deliberately eliminated the public noticing comment process.
Mr Clapton agreed that the lack of the comment process was deliberate as the government wanted to be able to execute the price controls as quickly as possible.
Dr Potts added that the nine vs 13 years really are affecting the R & D of small molecules for large populations and hopes to extend the period from nine to 13 years.
Mr Reilly believes that we should view this as the beginning of a troublesome law that could worsen over time. He pointed out that in a Commonwealth Fund podcast [20], it was mentioned that we need to exercise caution and establish some guardrails because we are uncertain about the potential extent of its impact on innovation. This issue revolves around an investment dispute between the industry and the Congressional Budget Office (CBO) regarding the number of new drugs that might be affected. It is crucial to engage with investors and industry experts since they are most knowledgeable about the likely impact. While they may not have precise information, they understand the need to protect themselves and adjust their behaviour accordingly. Ultimately, the outlook is grim, with a significant number of molecules and various indications expected to be adversely affected. Even those who support the law acknowledge that innovation and R & D will be negatively affected.
Question 2: Should we experiment with the IRA drug price negotiation approach to assess its potential in price control? If innovation is undermined, maybe we can find an alternative solution. Dr Potts noted that there are some great small molecules for lung cancer treatment. If we go back to year 2000, a drug called Tarceva, which was the very first epidermal growth factor receptor (EGFR) small molecule, was good but not great and had lots of side effects. However, from this, there was a staircase of investment and development that has led to continually better drugs for lung cancer treatment over the last 20 years. Some now have almost no side effects and allow for a great quality of life and longer life expectancies. We are also seeing similar staircases with pancreatic cancer, colon cancer and ‘melanoma drugs. However, what this IRA approach is saying is, lets eliminate these staircases and experiment with what might happen.
Mr Reilly added that Dr Potts had an interview conversation with Drs Fonseca and McAneny [12, 13], during which they discussed the system’s current state in terms of its research capabilities, highlighting that it is at its peak. The advantages in the US system before the implementation of IRA are recognized by Europe. Both the patient and provider communities were also in favour of the old system. Policymakers are struggling to make a logical argument for the benefits of the IRA system, which was proposed and pushed forward with little public input, as a system that has any benefits other than cost savings. The question remains: where do these cost savings accrue, and how do they manifest?
Dr Potts argued that when you consider a condition such as bladder cancer, the costs associated with the disease become substantial when the bladder has to be removed, along with the expenses for the drugs required afterwards. However, if this condition can be prevented with the development of a drug, these costs can be avoided. Unless you take into account the totality of what the drug is accomplishing, you cannot really quantify any savings.
Mr Clapton voiced his concern that there is a broader issue related to cost. From a legislative lawmaker perspective, he agrees with the long-term value that medicines offer, particularly either transformative or even curative therapies. However, unfortunately, the framework that the CBO uses is very narrow, primarily looking at short-term expenditures and not taking into account the long-term benefits to the healthcare system. In terms of the question, he noted that he is particularly concerned about the IRA. As the IRA price control mechanism moves forward, the baseline assumption for CBO is that more drugs are subject to negotiation, resulting in more saving for the federal government. This will make it increasingly challenging to unwind, especially when considering the fiscal challenges facing the US. For example, the Hospital Insurance trust fund which funds Medicare, is set to be introduced in a few years. The overall debt numbers are staggering, and this situation is only going to worsen, putting immense pressure on federal lawmakers to reduce spending. He added that unfortunately, if we are talking about repealing or even mitigating IRA, there will be a cost associated with it. And with each passing year, that cost will grow larger, making it increasingly difficult to repeal.
Question 3: How do we see the inter-relationship between the European marketplace and the US marketplace? Dr Olsen commented that in Europe, there have been changes and increased pressures, particularly on developers of innovative drugs. These pressures are now also present in the US, which does not bode well for innovation. In Europe, the World Health Organization regional office set up a stakeholder consultation called the ‘Access to novel medicines platform’, where discussions with governments took place. The idea of wealthier countries contributing more money for development was a topic of discussion. The pressures are quite apparent, and the concept of cost-sharing is conceivable.
Conclusions
The webinar provided important insight on key elements of the IRA’s Medicare drug price negotiation provisions, and the concerns surrounding their modification of successful, market-based models for delivering prescription medicines via a medical benefit (Medicare Part B) and pharmacy benefit involving competing prescription drug plans (Medicare Part D) that are strongly supported by beneficiaries.
Several fundamental flaws with the IRA’s drug pricing provisions surfaced, both in the construct of the statute and the approach CMS has taken to implementing it via sub-regulatory guidance. These flaws make clear that, despite its name, the programme is a form of government price setting, not negotiation, At the same time, the webinar brought to light several distinct flaws within the drug price negotiation programme that are not found even in European systems and are likely to worsen the harm done by the law in regards to continued progress and patient access to treatments and local physicians. These include extreme penalties for manufacturers failing to agree to negotiate, a basic lack of clarity and predictability in evidence standards and decision-making by CMS, lack of an open and transparent process by which patients and physicians can provide meaningful input, and a prohibition against appeals and judicial review.
In-depth discussions were held regarding the likelihood that the reforms would be unlikely to contain costs and would have a negative impact on innovation and patient access to new medicines. The role of European price setting was suggested as playing a role in the disparity of patient access and health outcomes between US patients and European patients. Due to the IRA’s ‘small molecule penalty’ and its initial application only in Medicare Part D, the statute’s damaging effects are most likely to be felt earliest and most acutely among US patients with cancer, chronic conditions like diabetes, cardiovascular disease, and immune-related conditions, and rare disease patients who are relying on current and future advances in small molecule medicines.
Acknowledgement
The Generics and Biosimilars Initiative (GaBI) wishes to thank all speakers and the moderator in delivering the presentations, implementing the panel discussion, and clarifying information when finalizing the meeting report, as well as Mr Michael S Reilly for his strong support through the offering of advice and information during the preparation of the webinar.
The authors would like to acknowledge the help of the webinar speaker faculty and all participants, each of whom contributed to the success of the webinar and the content of this report, as well as the support of the moderator in facilitating meaningful discussion during the panel discussions and contributing to the finalization of this meeting report.
Lastly, the authors wish to thank Ms Alice Rolandini Jensen, GaBI Journal ;Editor, in preparing and finalizing this meeting report manuscript.
Speaker Faculty, Panelists and Moderator
Speakers and Panelists Thomas R Barker, Esq, JD Charles M Clapton, Esq Matias Olsen Steven J Potts, PhD, MBA Michael S Reilly, Esq Professor Philip J Schneider, MS, FASHP, FASPEN, FFIPAndrew Spiegel, Esq
Moderator Steven Stranne, MD, JD
Editor’s comment
Speakers and moderator had provided feedback on the article content and panel discussion, read and commented the revised content of the manuscript, and approved the final report for publication.
Readers can watch replay of the IRA Medicare Drug Price Negotiations webinar via this link: https://youtu.be/1-JRK_mPXR0
Funding sources
The webinar was funded by ASBM.
The Alliance for Safe Biologic Medicines (ASBM) is a coalition of patient advocacy organizations, physicians, pharmacists, biopharmaceutical manufacturers, and others working to advance patient-centered health policy at the state, federal, and international level. Learn more at www.SafeBiologics.org
Competing interests: Mr Michael S Reilly, Esq is the Executive Director and employed by Alliance for Safe Biologic Medicines. Mr Reilly served in the US Department of Health and Human Services from 2002 to 2008.
Mr Thomas R Barker is a partner at the law firm of Foley Hoag, LLP where he represents multiple pharmaceutical, biological, and medical device manufacturers before the Centers for Medicare & Medicaid Services on matters relating to Medicare and Medicaid payment and coverage of their products. Mr Barker is also on the faculty of the George Washington University Schools of Law and Public Health and Health Services, and Suffolk University School of Law. He has no conflicts to report.
Mr Charles M Clapton is the Vice President, Federal Government Affairs, Gilead Science, Inc. Mr Clapton served as a Congressional staffer from 1995 to 2012, principally working for the House Energy and Commerce, Ways and Means and Senate Health, Education, Labor and Pensions (HELP) Committees.
Dr Steven J Potts is a consultant to a number of biotech drug development companies and serves on several biotech company boards as well as a volunteer position with the International Cancer Advocacy Network (ICAN).
Mr Andrew Spiegel is a patient advocate for the Global Colon Cancer Association as well as the World Patients Alliance. Both organizations receive funding from the pharmaceutical industry. Mr Spiegel also occasionally serves on advisory boards for the industry for which he receives some compensation. There are no conflicts of interest to report.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Michael S Reilly, Esq Thomas R Barker, Esq, JD Charles M Clapton, Esq Steven J Potts, PhD, MBA Andrew Spiegel, Esq
References 1. GaBI Online – Generics and Biosimilars Initiative. First drugs for Medicare price negotiation selected [www.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2023 Nov 6]. Available from: www.gabionline.net/policies-legislation/first-drugs-for-medicare-price-negotiation-selected 2. CMS.gov. Inflation reduction act and Medicare drug price negotiation [homepage on the Internet]. [cited 2023 Nov 6]. Available from: https://www.cms.gov/inflation-reduction-act-and-medicare/medicare-drug-price-negotiation 3. GaBI Online – Generics and Biosimilars Initiative. Four drugmakers sue the US government over the inflation reduction act [wwww.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2023 Nov 6]. Available from: www.gabionline.net/pharma-news/four-drugmakers-sue-the-us-government-over-the-inflation-reduction-act 4. GaBI Online – Generics and Biosimilars Initiative. More drugmakers sue over IRA yet one withdraws [wwww.gabionline.net]. Mol, Belgium: Pro Pharma Communications International; [cited 2023 Nov 6]. Available from: wwww.gabionline.net/policies-legislation/more-drugmakers-sue-over-ira-yet-one-withdraws 5. Gassull D, Bowen H, Schulthess D. IRA’s impact on the US Biopharma Ecosystem. Vital Transformation. 1 June 2023. Available from: https://www.bio.org/sites/default/files/2023-06/IRA%E2%80%99s%20Impact%20on%20the%20US%20Biopharma%20Ecosystem.pdf 6. University of Chicago. Philipson TJ, Durie T. The impact of HR 5376 on biopharmaceutical innovation and patient health. 29 November 2021. Available from: https://bpb-us-w2.wpmucdn.com/voices.uchicago.edu/dist/d/3128/files/2021/08/Issue-Brief-Drug-Pricing-in-HR-5376-11.30.pdf 7. No Patient Left Behind. 25 July 2022. The Honorable Chuck Schumer. Available from: https://nopatientleftbehind.docsend.com/view/e4cg7sgj6js5qenr 8. Potts S. Saving money for Medicare by abandoning new drugs for Medicare patients. Available from: https://rapport.bio/all-stories/ira-abandoning-new-cancer-drugs-for-medicare-patients 9. Potts S. Preserving the biotech social contract – we should all be pitching in. Available from: https://timmermanreport.com/2023/05/preserving-the-biotech-social-contract-we-should-all-be-pitching-in/ 10. Potts S. Measuring the damage: IRAs impact on small molecule drug development. NPLB. 31 March 2023. Available from:https://www.nopatientleftbehind.org/publications/ira-impact-on-small-molecule-development 11. Fishburn CS. IRA survey: biotechs bracing for impact. BioCentury. 16 March 2023. Available from: https://www.biocentury.com/article/647205/ira-survey-biotechs-bracing-for-impact 12. Rafael F, Potts S. KOL conversation. The IRA: How public policy impacts innovation. AZBio. Available from: https://www.azbio.org/how-public-policy-impacts-innovation 13. McAneny BL, Potts S. KOL conversation. The impact of the IRA on oncology patient care. AZBio. Available from: https://www.azbio.org/kol-conversation-the-impact-of-the-ira-on-oncology-patient-care 14. Pipes S. Europe negotiates a poor vaccine rollout; Forbes, April 2021. Available from: https://www.forbes.com/sites/sallypipes/2021/04/26/europe-negotiates-a-poor-vaccine-rollout/ 15. Wiggins J. Biosimilars Training Program 2023. Patient Advocate Perspective: Australia. [cited 2023 Nov 6]. Available from: https://youtu.be/HGjF8oBtzZw 16. IRA Patient Info [homepage on the Internet]. [cited 2023 Nov 6]. Available from. IRAPatientInfo.org 17. Smith: Democrat plan on drug costs will stifle innovation, San Antonio Express-News, 12 May 2021. Available from: https://www.expressnews.com/opinion/commentary/article/Smith-Democrat-plan-on-drug-costs-will-stifle-16172017.php 18. Spiegel A. Amend law to combat skyrocketing colorectal cancer rates (opinion). Reading Eagle. Available from: https://www.readingeagle.com/2023/05/13/amend-law-to-combat-skyrocketing-colorectal-cancer-rates-opinion/ 19. NICA, GCCA, PhRMA Litigation Asserts Price Setting Provisions in the Inflation Reduction Act are Unconstitutional. PhRMA. 21 June 2023. Available from: https://phrma.org/resource-center/Topics/Access-to-Medicines/Release-NICA-GCCA-PhRMA-Litigation-Asserts-Price-Setting-Provisions-in-the-Inflation-Reduction-Act-are-Unconstitutional 20. Seervai S. What the Inflation Reduction Act really means for health care. The Commonwealth Fund. 9 September 2022. Available from: https://www.commonwealthfund.org/publications/podcast/2022/sep/what-inflation-reduction-act-really-means-health-care
Author for correspondence: Michael S Reilly, Esq, Executive Director, Alliance for Safe Biologic Medicines, PO Box 3691, Arlington, VA 22203, USA
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Abstract: Undoubtedly the complexity of antibody drug conjugate (ADC) molecules where three components (monoclonal antibody, cytotoxic drug and appropriate linker) are involved poses a challenge for biosimilar development. As with all biomolecule development, both novel and biosimilar, it is important to choose orthogonal analytical techniques to interrogate Quality Attributes, particularly for higher order structure (HOS) studies and investigations into possible aggregates. Examples of suitable techniques are described.
Submitted: 27 June 2023; Revised: 1 August 2023; Accepted: 18 September 2023; Published online first: 2 October 2023
ADCs-The promise of precisely targeted drug delivery
Antibody drug conjugates (ADCs) are antibody-based anticancer therapeutics consisting of monoclonal antibodies attached to a cytotoxic drug via a linker of some kind. These highly targeted delivery systems offer the promise of lower cytotoxic drug levels in the patient and, as a result, potentially reduced drug side-effects [1].
The first globally approved ADC was Pfizer and Wyeth’s Mylotarg (gemtuzumab ozogamicin) in 2000, however, it was not until 2011 that the second ADC approval came in the form of Seagen and Takeda’s Adcetris (brentuximab vedotin). These early ADCs came with ‘black box’ warnings making clear that although the benefits outweighed the risks, there remained issues with toxicity. Over the next decade companies explored improved conjugation chemistries to link their non-proteinaceous drug to the antibody vector of choice with 14 ADCs in total receiving US Food and Drug Administration (FDA) approval to date, with many reaching blockbuster statuses.
The question is though, are ADCs a class of drug that may be almost immune to biosimilar competition given their complexity? Undoubtedly the development challenges are far greater which may limit the number of biosimilar sponsors willing to take them on. In 2021, the Drugs Controller General of India approved Zydus Cadila’s ‘similar biological’ of Genentech/Roche’s Kadcyla (trastuzumab emtansine) marketed as Ujvira for treating both early and advanced human epidermal growth factor receptor 2 (HER2) positive breast cancer. To date, neither FDA nor the European Medicines Agency (EMA) have yet approved a biosimilar ADC under their respective regulatory pathways.
Structural characterization and demonstrating biosimilarity of ADCs
The requirements for structural characterization of novel ADCs and potential biosimilar versions are the same as for other biomolecules in that the precepts of ICH Q6B [2] should be followed to achieve a full and detailed overall picture of primary and higher order structure (HOS). However, it must be noted that the requirement for conjugation and the nature of the conjugation chemistry itself mean that investigations need to be performed to:
determine the drug/antibody ratio (DAR)
locate the sites of attachment of the drug
look for reaction by-products attached in place of the drug, and
determine if any free linker is present on the ADC.
Careful analysis of conjugation products must be carried out to ensure that the conjugation itself has proceeded as expected. This includes determining that the antibody has not been adversely affected through, for example, oxidation, deamidation, non-specific drug binding or any other side reactions that are chemically feasible under the conjugation conditions used.
Many of the structural analyses performed will involve an analytical assessment of the primary structure, i.e. amino acid sequence, but it is important to remember that the attachment of drug groups with their own characteristics of size, charge distribution and hydrophobicity/hydrophilicity at various points could also influence secondary and tertiary structure, as well as creating the potential for aggregation.
For these reasons, it is important that the HOS is also investigated to fully understand the impact of the conjugation process on the monoclonal antibody (mAb) and the resultant ADC. It must be pointed out that alteration of the HOS between the parent mAb and the ADC is not necessarily in itself problematic, after all the ADC is not being marketed as a biosimilar to the parent mAb. Rather, the data are generated to give a better understanding of the product following conjugation. Furthermore, assessment of ADC HOS across different batches of product provides part of the structural characterization evidence for batch-to-batch consistency (or otherwise).
Alteration of the HOS of an ADC when compared to its native mAb may lead to questions regarding potential immunogenicity or could be a cause of increased aggregation, if observed. Furthermore, from a functional point of view, HOS changes may impact mAb target binding and therefore affect the ability of the mAb to deliver the drug to the site of action.
Analysis of secondary and tertiary structure
Secondary and tertiary structural techniques are used routinely in protein analysis, the most frequently used being circular dichroism (CD), fourier transform-infra-red (FT-IR), fluorescence analysis and nuclear magnetic resonance (NMR; both 1D (1 H) and 2D (1 H- 13 C) [3, 4]. These techniques are all equally applicable to ADCs since the protein structural features are preserved in ADCs, as would be expected. Each of these techniques works on a different principle and therefore will probe HOS in slightly different ways, resulting in unique outputs from each technique.
Circular dichroism Circular dichroism measures the differences in absorbance by the sample of right and left hand circularly polarized light. This technique works well for chiral molecules and, since proteins exhibit chirality, has proven effective for their analysis. Wavelengths used are in the far and near UV region, giving information on secondary and tertiary structure, respectively. The far UV data are computer processed and, with database searching (data obtained from analysis of proteins with known secondary structures), provide a breakdown of secondary structural features. The protein component of ADCs is just as amenable to CD analysis as the parent monoclonal antibody; thus, CD data can be generated allowing an investigation into the similarity, or otherwise, of the HOS structural profiles. The data shown in Figures 1 and 2 show the stacked near and far UV data, respectively, for several ADC batches and the parent mAb [unpublished results, 5]. All data images presented in this article are from these same ADC batches and parent mAb, analysed by different HOS techniques. The difference in the spectra in the region below 280 nm in Figure 1 is consistent with the difference in absorbance between the disulphides and the pair of free/conjugated thiols as a result of the conjugation chemistry (pers. comm).
FT-IR FT-IR uses infra-red light to investigate various bond flexures in the protein such as C=O, C-N, C-H, C-C and N-H. The chemical environments produced by the HOS features will have an impact on the precise wavelengths at which these flexures are seen. The absorption profile generated is subject to Fourier Transform mathematical interpretation and database interrogation to provide an output of relative abundances of secondary structural features. Figure 3 shows the FT-IR profiles of the same four ADCs and the parent mAb as shown in the previous figures [5].
It is very important to bear in mind that secondary structure analysis by different techniques will provide data that will reflect the sensitivity of that technique for particular secondary structural features. In other words, Far UV CD and FT-IR data, when processed, are unlikely to provide the same relative ratios of secondary structural features. This can be seen in Table 1 where the Far UV CD and FT-IR data for the four ADCs and parent mAb analysed in this study are compared. The fact that different values are obtained between these techniques is not in itself a problem since what matters is that batches compare to one another within the same technique and data set. The data are not being used to give a precise structural definition for the ADCs but are aiming to demonstrate similarity of result and therefore similarity of structure between batches.
Fluorescence Fluorescence analysis can be both intrinsic to the molecule and extrinsic by means of fluorophore addition to the sample. In the intrinsic sense, fluorescent analysis involves measuring the fluorescence profile of tyrosine and tryptophan residues in the molecule using a spectrophotometer. The profile generated is related to the chemical environments that these amino acids find themselves in and thus the fluorescence profile gives information about the overall nature of the Tyr and Trp chemical environments. Relative amounts of secondary structural features are not determined with this technique. An example of intrinsic fluorescence is given in Figures 4 and 5 below for the same ADC batches and parent mAb as shown in the previous figures [5].
1D and 2D NMR NMR relies on measuring the response of magnetic nuclei in the sample (most commonly 1 H and 13 C) to a radio frequency pulse applied perpendicular to a constant magnetic field. As the magnetic nuclei relax back to their ground state following the RF pulse they emit electromagnetic radiation, giving rise to the NMR signal. Again, the nature of the chemical environment that these atomic nuclei find themselves in will influence their relaxation profiles and this in turn influences the NMR profile.
A larger amount of structural information can be obtained by monitoring 2 magnetic nuclei in one experiment &ndash a so-called 2D NMR analysis. Most commonly 1 H and 13 C are measured but 1 H and 15 N measurements can also be used (although the relative abundance of 15 N is lower than 13 C). This gives rise to an intricate pattern of signals that are a function of the local environments the 1 H and 13 C nuclei find themselves in.
For ADCs and monoclonal antibodies, NMR particularly in the 2D sense, can be challenging due to the large size of the molecule, structural flexibility and long correlation time. Nonetheless, there are techniques available that allow data to be generated on such large proteins. Figure 6 gives an example of 1D NMR data generated for the same four batches of ADCs and the parent mAb as shown in previous figures (unpublished data).
Since these techniques are used to assess the HOS of a molecule, it is critical to analyse the unconjugated antibody alongside the ADC to demonstrate what to expect from the data from the mAb’s native state and to investigate if any changes have occurred in the HOS as a result of the conjugation process. One other point to consider is that each of these techniques described above will interact with the sample as the physics of the technique demands. Thus, since each technique works from different physical principles it follows that the outputs will be different between the techniques. This is perfectly fine from a structural characterization point of view and is a very good example of orthogonality, where data from different analytical approaches are used to support conclusions from the various techniques, forming a more self-sustaining whole.
The idea of orthogonality in HOS analysis is just as applicable for ADCs as it is for other proteins, since each individual technique will have its own sensitivities for different structural features such as CD having greater sensitivity for alpha helices compared to FT-IR. Any perturbation of the HOS in the ADC may disrupt these motifs to a greater or lesser degree both locally to the site(s) of conjugation or more globally across the molecule, therefore techniques need to be as all-encompassing as possible.
It is important to bear in mind that the ADC drug and linker will also have their own spectroscopic profile. Depending on the drug, it may have absorption characteristics in the spectral range of some of the instrumentation (CD in particular). A spectroscopic profile of the native drug is therefore absolutely necessary when assessing HOS data from an ADC to help assess any impact the drug is having on the overall ADC profile. Thus, comparison of the drug, ADC and native mAb can explain features seen in the ADC absorbance profile.
Aggregation
The chemical processes used to produce ADCs could result in enhanced aggregation as a result of structural disruption of the mAb and this needs to be investigated. Again, a sample of the native mAb should be analysed alongside the ADCs to provide baseline aggregate levels.
Just as with HOS analyses, orthogonality is very important in aggregation studies. This comes out of the general principle of the value of orthogonal investigations, as recognized by the regulatory agencies, stemming from the fact that different aggregation techniques will give different values for the level of aggregation due to the nature of the techniques themselves. A combination of sedimentation velocity ultra-centrifugation (SV-AUC) and size exclusion chromatography with multi-angle laserlight scattering (SEC-MALS) for aggregation studies have been shown to be applicable to ADCs. Data from the SEC-MALS and SV-AUC analyses of the same four ADC batches and the parent mAb as shown in previous Figures are shown in Figures 7 and 8, respectively (unpublished results). The parent mAb is likely to have slightly different hydrodynamic properties to the ADCs and this will result in slightly different sedimentation coefficients.
Discussion: HOS analysis – a necessity for ADC development
The range of HOS techniques available and applicable to structural analysis of biopharmaceutical products are equally applicable, and indeed vital, for the structural characterization of ADCs in order to gain a full structural understanding of the product. The fact that these techniques all have different biophysical attributes and thus analyse HOS from different perspectives is of great significance for building up a strong picture of biopharmaceutical HOS. When these techniques are used in an orthogonal manner, the various different features of individual techniques such as CD, FT-IR and NMR, with their unique biophysical properties, serve as meaningful molecular probes across different aspects of structure. Together with comparative data for the parent mAb, evaluations can be made not only with regard to batch-to-batch comparability for the ADC or in an ADC biosimilarity study but also for assessment of change in HOS compared to the parent mAb. This knowledge can be relevant for understanding any impact in terms of function or possibly aggregation that may be seen with the ADC. Furthermore, investigations of the extent of structural change between parent mAb and ADC can lead to the investigation of new conjugation chemistries that have less impact on HOS, thus producing mAb drug scaffolds more closely aligned structurally to the parent mAb.
Orthogonality of protein analytical aggregation techniques can also be successfully applied to ADCs with the data being used to either confirm satisfactory levels of aggregation or suggest possible modifications to process/purification procedures if levels are outside an expected or acceptable range. Thus, aggregation studies of ADCs are inextricably linked to HOS investigations and product development.
Conclusion
In summary, if ADCs are to become targets for biosimilar development, then, as with all biosimilar structural studies, their HOS and propensity to aggregate needs to be investigated using appropriate techniques such as those described here.
Funding sources
This paper is funded by BioPharmaSpec Ltd (www.biopharmaspec.com).
Competing interests: Dr Richard L Easton joined BioPharmaSpec in 2016 as Technical Director for Structural Analysis and is responsible for management of all aspects of carbohydrate and glycoprotein characterization at the primary structure level.
Provenance and peer review: Not commissioned; externally peer reviewed.
References 1. Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021;18(6):327-44. 2. European Medicines Agency. ICH Q6B Specifications: test procedures and acceptance criteria for biotechnological/biological products – Scientific guideline [homepage on the Internet]. [cited 2023 Aug 1]. Available from: https://www.ema.europa.eu/en/ich-q6b-specifications-test-procedures-acceptance-criteria-biotechnological-biological-products 3. Astier A. Importance of the determination of the higher order structure in the in-use stability studies of biopharmaceuticals. Generics and Biosimilars Initiative Journal (GaBI Journal). 2020;9(2):49-51. doi: 10.5639/gabij.2020.0902.009 4. Vieillard V, Astier A, Paul M. Extended stability of a biosimilar of trastuzumab (CT-P6) after reconstitution in vials, dilution in polyolefin bags and storage at various temperatures. Generics and Biosimilars Initiative Journal (GaBI Journal). 2018;7(3):101-10. doi: 10.5639/gabij.2018.0703.022 5. McKee C, Chapman C, Bayley C. SDE-100 a stochastic cysteine linked vedotin ADC: assessing comparability of higher order structure using multiple orthogonal analytical approaches. Results presented as poster at 13th World ADC; 13&ndash16 March 2023; London.
Author: Richard L Easton, BSC (Hons), DIC, PhD, Technical Director – Structural Analysis, BioPharmaSpec Ltd, Suite 3.1 Lido Medical Centre, St Saviour, Jersey, JE2 7LA
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
Author byline as per print journal: Ka-Liong Tan1, DPhil; Ainoon Othman1; Irwan Mohd Subri2,3; Noor Fadzilah Zulkifli1; Mohd Mahyeddin Mohd Salleh3; Nazariyah Yahaya4; Khairun Nain Nor Aripin1; Shahirah Nadiah Shaharuddin5; Seri Azalina Mohd Ghazalli6; Muhammad Syazan Sulaiman6
Abstract:
In recent years, there has been a rapid growth of the halal pharmaceutical industry, especially in the supply chain of solid oral dosage forms of medication. This article outlines aspects of the Halal Management System (HMS) in the development and production of halal pharmaceuticals. It explains the needs and requirements of HMS and identifies the challenges faced in implementation. The article outlines aspects of execution and hurdles encountered when standardizing halal certification. The article also highlights the need for systematic traceability systems and effective product recall mechanisms to ensure adherence to halal requirements. It also highlights the grey areas for halal in terms of pharmaceutical manufacture that are brought about by use of non-halal raw materials, e.g. alcohol, gelatine, glycerin, lecithin, glutamic acid and stearates.
Submitted: 4 April 2023; Revised: 22 August 2023; Accepted: 23 August 2023; Published online first: 5 September 2023
Implementation of Halal Management System (HMS) in the manufacturing of solid oral dosage forms
Halal is an integral observance for all Muslims. This concept originates from an Arabic word and can be defined as permissible by shariah law. The consumption of halal foods is mandated under Islamic teachings and includes, water and beverages, meals and snacks, as well as pharmaceutical medicines. Medicines also fall under Islamic dietary law and are required to be halal and Muslims are forbidden to use illicit drugs except in an emergency [1].
In recent years, the global demand for halal pharmaceuticals has been increasing. This comes with the growth of the global population of Muslims which is expected to grow from 1.6 billion to 2.2 billion by 2030 [2]. It is estimated that halal pharmaceuticals now account for 22% of the total value of all halal products [3]. Furthermore, halal pharmaceuticals have also gained increasing acceptability among non-Muslims due to ethical consumption issues such as social responsibility, stewardship, economic and social justice, animal welfare, as well as ethical investment. In addition, many pharmaceutical companies are aspiring towards bigger investment and development of halal-certified products.
The term pharmaceutical refers to both prescription and non-prescription medicinal products in finished dosage forms, i.e. biopharmaceuticals, radiopharmaceuticals, and traditional medicines. The dosage forms can be administered via oral intake, through body orifices, given as injections, implants, or through topical application [4]. Common orally administered drugs can be in the forms of tablets, soft-gel capsules, chewable, orally disintegrating tablets, sublingual tablets, capsules, lipids, and powder. Table 1 shows the classification of oral dosage forms available on the market. According to a recent survey, oral dosage forms are the most popular means of delivering active pharmaceutical ingredients (API) to patients [5]. Oral dosage forms provide better protection against moisture, oxygen, and light before the medication is consumed and released into the body as compared to injectables and topical formulations. Halal pharmaceuticals are better outlined in the supply chain and Table 2 lists the halal-related standards available in Malaysia.
The HMS is an approach used in the detection of non-halal contaminations which incorporates control steps to the production process to ensure that products and services are halal and safe. HMS is crucial to regulate non-halal elements and safeguard the integrity of halal products and services. It covers all aspects of sourcing, manufacturing procedure, packaging, and logistics, see Figure 1. The system provides both guidelines for hazard analysis critical control point (HACCP) and good manufacturing practice (GMP) in product processing to ensure compliance with shariah requirements. Overall, when it comes to halal pharmaceutical production, processes must adhere to local and international standards including: GMP, good hygiene practice (GHP), good clinical practice (GCP), good laboratory practice (GLP), good storage practice (GSP), good engineering practice (GEP), and good distribution practice (GDP). In view of the complexity of HMS, more efforts are required for stakeholders and regulators of the pharmaceutical industry worldwide to understand these features and ensure the adherence of halal-pharmaceuticals.
Medicines are often prescribed in emergencies to treat conditions that may be life-threatening, thus the issue of whether the product is halal issue is not always the highest priority. This can partly explain why the halal pharmaceutical industry remains in a nascent stage. In the absence of strong market demand, halal pharmaceuticals need to rely on governments and industry players in Muslim-dominant countries to promote awareness. Suppliers fail to prioritize the halal certification of pharmaceuticals and, to address this challenge, some experts recommend that governments and manufacturers implement the relevant halal management and accreditation processes themselves.
To implement halal management, continuous social science research, clinical experiments, and development initiatives are necessary to identify alternatives to non-halal ingredients. Established international halal regulations are now gaining more recognition, especially those concerning oral dosage forms.
The growth of the halal supply chain to produce solid oral dosage pharmaceuticals presents various challenges. Despite halal’s increasing popularity by both Muslims and non-Muslims, there is a lack of understanding about the halal concept, leading to a poor recognition of its requirements in pharmaceutical preparation. The complexity of ingredients and processes involved makes the implementation of HMS challenging. Non-compliance with halal regulations can result in product recalls, emphasizing the need for systematic traceability and resource utilization to minimize costs during such a crisis. Furthermore, the lack of Muslim human capital in the industry poses an additional obstacle to sustainable HMS implementation. Therefore, there is a need to review the disputes and challenges faced by the implementation of HMS, focusing on the manufacturing of solid oral dosage forms.
This article aims to provide justification for the implementation of HMS in the manufacturing of solid oral dosage forms by the pharmaceutical industry. The article also provides some solutions and strategies to improve compliance with halal regulations.
Standardizing halal certification
International organisations, such as the World Halal Council (WHC) and the Standards and Metrology Institute for Islamic Countries, work together to oversee the standardization of halal certification and accreditation processes. Here, stakeholders actively participate in the development process through working groups and public comments [6]. Several countries, including South Korea and ASEAN (Association of Southeast Asian Nations) have set up national halal certification policies in accordance with International Standards Organization (ISO). The development process also complies with the guidelines established by the World Trade Organization (WTO). Table 3 lists the halal certification authorities in Southeast Asia.
HMS is the primary industry standard used to maintain the halal status of products. Table 4 depicts the structure of HMS. The halal procedure is key to ensure that all halal products are produced responsibly [7]. The HMS clauses are established to manage the overall quality of an organization in accordance with halal requirements [8]. The certification process may vary based on individual national policies. In Malaysia, medium and large industries can only get halal certification by implementing the complete Halal Assurance System (HAS) [9]. However, small and micro enterprises can obtain the certification via the creation of an Internal Halal Committee (IHC) in accordance with the Malaysian HMS and the Manual Procedure for Malaysian Halal Certification [10-12]. IHC is responsible for designing, monitoring, and assuring the implementation of the six principles of HAS as shown in Table 5. It should consist of at least four members, namely two Muslims at the managerial level, one involved in the acquiring and sourcing process, and the halal executive who is responsible for monitoring the halal affairs of the company [13]. Figure 1 shows an example of the IHC composition.
Regarding halal pharmaceuticals, it is key that guidelines are present to standardize the halal requirements. In Malaysia, this guideline (MS 2424:2019 Halal pharmaceuticals &ndash General requirements) was developed for the production and handling of halal medicines to ensure that pharmaceutical companies comply with shariah requirements. Furthermore, the manufacturers must also establish a dedicated processing line for halal pharmaceuticals.
Figure 3 illustrates the steps of HAS implementation. All materials applied in the manufacturing of halal pharmaceuticals, including API, must comply with halal principles. Materials acquired from suppliers under contracts, any other commercial arrangement, or made in-house must also be subjected to the same requirement. More importantly, all materials must be najs-free, i.e. halal (as described in section 4.0). ‘Najs’ is an Arabic term that means ‘filth’ and is considered non-permissible for consumption according to Islamic law. Finally, companies must use dedicated vehicles with appropriate hygiene and sanitation condition for the transportation of all medicines.
It is key to highlight that, to achieve stainable production of halal pharmaceuticals, manufacturers should employ management and operational team personnel to monitor, identify, record, and report any problems in the halal process based on international standards. This can minimize the risk of contamination, mix-ups, and errors in the production processes, thus protecting consumers from potential risks of sub-standard medicines. Pharmaceutical manufacturers applying for halal certification for their products should comply with all the safety and hygiene requirements and adhere to the requirements of shariah law. Dedicated equipment must be used to avoid cross-contamination of halal by non-halal products [14]. In the cleansing procedure, the equipment must be cleaned using clay to remove any microbial contamination, followed by washing, spraying, and rinsing. Segregation of halal and non-halal products at every stage is obligatory, including storing, displaying, and transporting [15]. Furthermore, primary packaging materials must be customized to prevent contamination post-production. The origin and nature of the paper or plastic packaging, inks, films, and glue are also of concern for halal status. On the packaging, information incorporating the name, brand, minimum content in metrics, name and address of manufacturer/distributor, list of ingredients, code number representing production batch, manufacturers, as well as expiry dates must be outlined.
Non-compliance with halal standards and need for a reliable traceability system
With the increasing interest in the halal pharmaceutical market, several problems have emerged, such as non-compliance with halal standards and shariah law and the fraudulent use of halal logo and terms. For instance, a halal product was found to be contaminated with non-halal content, i.e. a pig’s DNA, and certified using a fake halal logo, resulting in the suspension of halal certification for the product [16]. This highlights the importance of a credible system to safeguard the integrity of halal products. For the pharmaceutical industry, a systematic traceability system used by manufacturers is essential to sustain HMS. Table 6 lists the known halal non-conformance cases in pharmaceutical establishments. These incidents are commonly detected by authorities when conducting audit checks.
The halal traceability framework was established to maintain the integrity of halal products and ingredients throughout the production and supply chain [17]. The IHC and HAS been put in place to facilitate recall procedures for any products that have been recognized as non-compliant [17]. In Malaysia, the National Pharmaceutical Regulatory Agency (NPRA) regulates halal products, manufacturing plants, and work methods, to expand the production and supply of halal products for the global market. The traceability HMS requirement is in tandem with the existing international standards, i.e. the Pharmaceutical Inspection Co-operation Scheme (PIC/S) GMP Guide. This document is also adhered to by the NPRA. The existing traceability system requirements outlined in the PIC/S standard facilitate the implementation of HMS. With outlined traceability practices in place, minimal integration is needed for the adoption of the HMS guideline. Table 7 compares the traceability for HAS, HACCP and GMP.
Product recall management is the final step in traceability systems. In general, this refers to the process of removing defective medicines from the supply chain, ideally before they reach consumers. For example, these will be products that may cause illness or harm, i.e. unsafe food products, products with potentially adverse effects, or contaminated pharmaceutical products. All manufacturers are required to have effective product recall management systems in place to ensure consumer safety. This is also key for halal products and for ensuring their halal status. A product recall can damage the reputation and financial standing of a company. As a damage control strategy, companies must have a good recall management system in place that include checks for halal compliance.
During a recall crisis, a systematic traceability system and efficient utilization of resources can cut down cost. For example, an individual transportation network is vital to reduce transportation costs and to ensure the halal status during transportation. Overall, preventing the occurrence of product recalls will lead to a high level of customer confidence in halal integrity in the halal industry, particularly in terms of halal assurance.
Halal disputes and proposed solutions
Overall, there is increasing awareness among consumers, health experts, and various organizations of the need for solid oral dosage formulations that are safe, efficacious, high quality, hygienic, and compliant with religious obligations. More pharmaceutical manufacturers are exploring new values in their production process, especially in terms of identifying suitable alternatives that can substitute non-halal ingredients in the process of halal pharmaceutical production, see Table 8.
To achieve that, comprehensive scientific knowledge of all aspects of pharmaceuticals, including production, transport, and storage is a prerequisite to support halal requirements. Ideally, pharmaceuticals should be developed through various research and development programmes that will facilitate the creation of more alternatives for non-halal ingredients in halal pharmaceutical production. These include intoxicants, pork and its by-products, the meat of dead animals, and blood. Muslims are also prohibited from consuming animals that are grouped as carnivores and predatory birds such as dogs, tigers, owls, and hawks. These ingredients are termed &ndash najs and are thought to be ritually unclean. Despite the criteria for halal being fairly clear, halal pharmaceuticals do present some grey areas in terms of manufacturing processes. Below is a summary of a number of components of pharmaceuticals that are not halal compliant.
Alcohol The use of alcohol in producing API. Although the use of ethanol derived from the manufacturing of alcoholic beverages is not allowed, the use of alcohol compounds as processing aids and stabilising agents is permissible as its trace amount in the final product (0.01%v/v) will not be intoxicating [18].
Gelatine A long-standing issue affecting Muslim consumers is the use of gelatine from pigs (porcine) and cows (bovine) that are not slaughtered according to Islamic shariah law [19]. With the rise in demand as well as religious concerns surrounding gelatine, there is a need to search for affordable, abundant, and sustainably accessible alternatives [20].
One potential alternative to conventional gelatine (non-halal porcine and bovine) is to ensure that gelatine is derived from animal waste produced in Muslim countries [21]. It is estimated that about 24% of all gelatine originates from bovine and other cattle waste products [22]. Ovines, i.e. sheep and goats are other mammalian sources to extract gelatine [23]. In addition, gelatine derived from chicken by-products can also be considered and used as there is a large amount of chicken waste that includes chicken skin, bones, and shanks. Lastly, fish-sourced gelatine is acceptable to people of all religions.
Glycerine Glycerine, also referred to as glycerin or glycerol, is used in various products including cosmetics, pharmaceuticals, and foods. Glycerine sourced from animal fat needs to be regulated under the shariah rulings of slaughters to be considered halal. It is not acceptable if the ingredient is extracted from animals that are alive. On the other hand, Halal glycerine can be derived from plants such as palm oils and soybeans.
Lecithin Lecithin is a fatty substance that occurs naturally in the body tissues of humans, animals, and plants. It can be found naturally in soybeans and yolk. However, it is considered non-halal if obtained from the fatty tissues of non-slaughtered animals. Lecithin functions as an emulsifier by suspending fats and oils to prevent mixing with other substances. It has a variety of medicinal and commercial uses with extensive health benefits. Lecithin supplements are often prescribed to supplement the treatment of high cholesterol, ulcerative colitis, and Alzheimer’s disease.
Glutamic acid Glutamic acid is an α-amino acid involved in the production of proteins of all living things. Pharmaceutically, glutamic acid supplements have been used to treat behavioural problems and as a supportive treatment of cognitive diseases. It has also been prescribed to prevent nerve damage in chemotherapy patients. In addition, it is also widely used in a variety of cosmetic products.
Glutamic acid can be found naturally in poultry, fish, and all high-protein foods. However, glutamic acid from non-slaughtered animals and pigs are not acceptable for use in halal products.
Disputes of the use and non-halal nature of these components of pharmaceuticals have sparked disputes and constructive discussions in the industry for a long time. The labelling of gelatine, glycerine, lecithin is compulsory to ensure its source of origin is clearly stated. The relevant authorities ensure that all related activities for the manufacturing and handling of halal pharmaceuticals are properly recorded. All businesses must maintain halal control points to ensure that chemicals, reagents, equipment, and other necessities are approved as halal [24]. To date, the scarcity of information remains a key constraint when it comes to the global halal pharmaceutical industry. Most suppliers are not aware of the opportunities in local and global markets. They are also not well versed in the importance of halal certification. In addition, most consumers also have a low level of concern about the content of medications and their halal status. Currently, there is no obligation for the clinics or pharmacy departments to label the micro packaging of medicine. As such, Muslim patients who are not informed of the presence of non-halal materials in the drugs would have unknowingly consumed the prescribed medicinal goods instead of sourcing halal substitutes of the same medicines. This factor contributes to the narrow exploration of the halal market and the difficulty that pharmaceutical manufacturers face in sourcing halal ingredients. The creation of a larger demand from the Muslim population is essential to sensitize the involved stakeholders and push them to undertake more halal product development.
Moreover, at present, the lack of human capital continues to act as a deterrent to the development of the halal pharmaceutical industry. The production of medicines with desirable pharmaceutical qualities that also satisfy religious obligations requires the participation of all involved parties in the manufacturing process. While the halal pharmaceutical industry is growing globally, there is still a lack of understanding of the halal concept, with most of the population associating it with religious matters. Even though religion plays a significant role in halal pharmaceuticals, more awareness about halal products should be instilled to facilitate their widespread production and uptake. Halal education programmes should be developed to educate the producers and the public about the role of halal pharmaceuticals in providing healthy, hygienic, and safe pharmaceutical drugs.
Conclusion
This article has outlined that HMS is an extension of HACCP and GMP guidelines in oral dosage processing to ensure compliance with shariah requirements. It highlights that it is important for manufacturers to uphold their moral commitments and safeguard the concerns of consumers when it comes to halal. This will allow local and international halal commerce to flourish. Non-compliance with halal regulations can result in oral dosage product recall. To reduce the cost during recall, a systematic traceability system is vital. Good HMS implementation can assure the quality and safety of oral dosages to embolden the trust and confidence of patients toward halal pharmaceutical products.
Acknowledgements
Contributors: We thank Maria Arshad for technical editing the manuscript for grammar and syntax. This manuscript underwent proofreading service by Proofreading by A UK PhD (Registration: NS0163592-K).
Funding sources
This work was financially supported by the Duopharma R&D fund (A2-5-21-804111-16).
Disclosure
KL Tan, A Othman, I Mohd Subri, NF Zulkifli, MM Mohd Salleh, N Yahaya, and KN Nor Aripin are affiliated to Universiti Sains Islam Malaysia (USIMs). SN Shaharuddin is affiliated to Kolej Universiti Islam Perlis (KUIP). All received consultation fee from Duopharma Biotech Berhad, Malaysia.
Competing interests: The authors have no declared conflicts of interests.
Provenance and peer review: Not commissioned; externally peer reviewed.
Authors
Ka-Liong Tan1 Ainoon Othman1 Irwan Mohd Subri2,3 Noor Fadzilah Zulkifli1 Mohd Mahyeddin Mohd Salleh3 Nazariyah Yahaya4 Khairun Nain Nor Aripin1 Shahirah Nadiah Shaharuddin5 Seri Azalina Mohd Ghazalli6 Muhammad Syazan Sulaiman6
1Faculty of Medicine and Health Sciences, University Sains Islam Malaysia (USIM), Persiaran Ilmu, Putra Nilai, 71800 Nilai, Negeri Sembilan, Malaysia 2Institute of Fatwa and Halal (IFFAH), University Sains Islam Malaysia (USIM), Persiaran Ilmu, Putra Nilai, 71800 Nilai, Negeri Sembilan, Malaysia 3Faculty of Syariah and Law, University Sains Islam Malaysia (USIM), Persiaran Ilmu, Putra Nilai, 71800 Nilai, Negeri Sembilan, Malaysia 4Faculty of Science and Technology, University Sains Islam Malaysia (USIM), Persiaran Ilmu, Putra Nilai, 71800 Nilai, Negeri Sembilan, Malaysia 5Faculty of Islamic Studies, Kolej Universiti Islam Perlis (KUIPs), Kuala Perlis, 02000, Perlis. 6Duopharma Biotech Berhad, Suite 18.06, Level 18, CIMB HUB, No. 26, Jalan Sultan Ismail, 50250 Kuala Lumpur
References 1. Al-Fatih S, Esfandiari F. Halal food in South East Asia: are we looking forward? 2020: 166-169. doi: 10.2991/aebmr.k.200226.034 2. Norazmi MN, Lim LS. Halal pharmaceutical industry: opportunities and challenges. Trends Pharmacol Sci. 2015:36(8):496-7. 3. Mohezar S, Zailani S, Tieman M. Tapping into the halal pharmaceutical market: issues and challenges. In: Contemporary issues and development in the global halal industry. Selected Papers from the International Halal Conference 2014. Springer Singapore. 2017. p. 531-41. 4. Zarif MM, Yusof AF. The use of forbidden materials in medicinal products: an Islamic perspective. Middle-East Journal of Scientific Research 13. Approaches of Halal and Thoyyib for Society, Wellness and Health. 2013. doi:10.5829/idosi.mejsr.2013.16.s.10022 5. Markl D, Zeitler JA. A review of disintegration mechanisms and measurement techniques. Pharm Res. 2017:34(5):890-917. 6. Jais AS. Halal related Malaysian standards. Halal Note Series-Halal Common. 2019;1. 7. Perdani CG, Chasanah NU. Evaluation of halal assurance system (HAS) implementation on bakery products processing in small and medium enterprises (case study in X Bakery Batu, East Java). IOP Conference Series: Earth and Environmental Science. 2018:131(1):012023. 8. Evans JR, Lindsay WM. The management and control of quality. Thomson/South-Western; 2005. 9. Idris I, Alias SS, Singh SK. Perception of muslim consumers towards halal branding in advertising. Int J Criminol Sociol. 2020. 10. Jamaludin MA. Fiqh Istihalah.integration of science and Islamic law. Revelation and Science. 2012;2(2):117-23. 11. Salleh AS, Romli F, Salleh KM, Adnan A. Role of Internal Halal Committee in ensuring business sustainability: the case of a multinational slaughterhouse. J Bus Manag Account. 2020:10(2):57-65. 12. Abdullah MS, Noordin MI, Ismail SI, Mustapha NM, Jasamai M, Danik MF, et al. Recent advances in the use of animal-sourced gelatine as natural polymers for food, cosmetics and pharmaceutical applications. Sains Malaysiana. 2018;47(2):323-36. 13. Riaz MN, Chaudry MM. General guidelines for halal food production. In: Handbook of halal food production. CRC Press; 2018. p. 17-28. 14. Di Foggia G, Ferrari S, Lazzarotti V, Pizzurno E. Innovation process for Halal product development: an empirical analysis of Italian firms. Manag Res Pract. 2011: 3(1):27-47. 15. Lam Y, Alhashmi SM. Simulation of halal food supply chain with certification system: a multi-agent system approach. In: Intelligent Agents and Multi-Agent Systems: 11th Pacific Rim International Conference on Multi-Agents. Berlin, Heidelberg: Springer; PRIMA 2008; p. 259-266. doi:10.1007/978-3-540-89674-6_29 16. Majdina Nordin FN, Jasimah Wan Mohamed Radzi CW. Religion and cosmetics: guidelines for preparing products aimed at the Muslim world based on the interpretation of halal cosmetics in Malaysia. J Cosmet Sci. 2021;72(2):139-54. 17. Zainuddin N, Saifudin AM, Deraman N, Osman AA. The effect of halal traceability system on halal supply chain performance. Int. J Sup. Chain Mgt. 2020;9(1):490-8. 18. Maizirwan M, MS Hamzah. Halal issues in pharmaceutical products: urgent need to have modern and efficient production of pharmaceuticals and biopharmaceuticals. Halal Pages. 2010:56-63. 19. Morrison NA, Clark RC, Chen YL, Talashek T, Sworn G. Gelatin alternatives for the food industry. In: Nishinari K, editors. Physical chemistry and industrial application of gellan gum. Berlin, Heidelberg: Springer; 1999. p. 127-31. 20. Alzeer J, Hadeed KA. Halal certification of food, nutraceuticals, and pharmaceuticals in the Arab world. Handbook of healthcare in the Arab world. 2021:765-87. doi:10.1007/978-3-319-74365-3_36-1 21. Alao BO, Falowo AB, Chulayo A, Muchenje V. The potential of animal by-products in food systems: production, prospects and challenges. Sustainability. 2017:9(7):1089. 22. Uddin SM, Hossain MM, Sagadevan S, Al Amin M, Johan MR. Halal and Kosher gelatin: Applications as well as detection approaches with challenges and prospects. Food Bioscience. 2021:44:101422. 23. Abdullah MS, Noordin MI, Ismail SI, Mustapha NM, Jasamai M, Danik MF, et al. Recent advances in the use of animal-sourced gelatine as natural polymers for food, cosmetics and pharmaceutical applications. Sains Malaysiana. 2018;47(2):323-36. 24. Department of Islamic Development Malaysia (Jabatan Kemajuan Islam Malaysia), Malaysia Halal Management System (MHMS) 2020 [homepage on the Internet]. [cited 2023 Aug 22]. Available from: https://smarthalal.com.my/manual.php
Author for correspondence:Ka-Liong Tan, DPhil, Faculty of Medicine and Health Sciences, University Sains Islam Malaysia (USIM), Persiaran Ilmu, Putra Nilai, 71800 Nilai, Negeri Sembilan, Malaysia
Disclosure of Conflict of Interest Statement is available upon request.
Permission granted to reproduce for personal and non-commercial use only. All other reproduction, copy or reprinting of all or part of any ‘Content’ found on this website is strictly prohibited without the prior consent of the publisher. Contact the publisher to obtain permission before redistributing.
GaBI Journal is an independent and peer reviewed academic journal. GaBI Journal encompasses all aspects of generic and biosimilar medicines development and use, from fundamental research up to clinical application and policies.