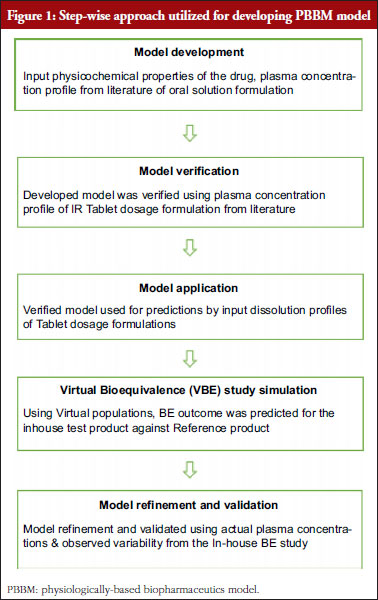
Real-world evaluation of dosing and haemoglobin A1c (HbA1c) in patients converted from insulin glargine (Lantus®) to insulin glargine (Basaglar®) in Saudi Arabia
Introduction/Study objective: About 28% of Saudi Arabia population above 15 years old are diagnosed with diabetes. Basal insulin glargine Lantus and its’ biosimilar Basaglar, are long-acting insulin that regulate blood glucose levels over 24 hours. Patients switch between basal insulins for various reasons including adverse events, availability and cost. Switching should be done with careful […]